Resumen
Introducción. La poliposis adenomatosa familiar (PAF) es una enfermedad autosómica dominante hereditaria causada por una mutación en el gen APC (adenomatous polyposis coli). Su sintomatología inicia de forma temprana y el riesgo de desarrollar cáncer colorrectal (adenocarcinoma) es casi del 100% si no se recibe tratamiento oportuno. Al ser una enfermedad infrecuente con alta morbimortalidad, es pertinente hacer estudios al respecto.
Presentación de los casos. Se presenta el caso de una PAF registrada en dos hermanos adolescentes que se manifestó con hemorragia digestiva. En ambos pacientes se realizó colectomía por laparoscopia y descenso endorrectal ileoanal más ileostomía y la evolución postoperatoria fue favorable y sin complicaciones. Los dos hermanos continuaron en controles ambulatorios por el servicio de cirugía pediátrica.
Conclusiones. La PAF es una enfermedad poco frecuente en población pediátrica. La colectomía es el tratamiento de elección, ya que con esta se controlan los síntomas y la morbimortalidad asociada. La evaluación genética es fundamental para hacer un adecuado acompañamiento en la toma de decisiones a corto, mediano y largo plazo en pacientes con esta enfermedad.
Abstract
Introduction: Familial adenomatous polyposis (FAP) is an inherited autosomal dominant disease caused by a mutation in the APC (adenomatous polyposis coli) gene. Symptoms appear early, and the risk of developing colorectal cancer (adenocarcinoma) is almost 100% if not treated promptly. As it is a rare disease with high morbidity and mortality, it is pertinent to carry out studies on this condition.
Case presentation: This is a report on FAP in two adolescent siblings that presented with gastrointestinal bleeding. Both patients underwent colectomy by laparoscopy and transanal endorectal pull-through plus ileostomy, and their postoperative progress was favorable and without complications. Both siblings received outpatient follow-up by the pediatric surgery service.
Conclusions: FAP is a rare disease in the pediatric population. Colectomy is the treatment of choice, since it controls symptoms and the associated morbidity and mortality. Genetic assessment is essential to provide adequate support in decision-making in the short-, medium- and long term in patients with this disease.
Introduction
Familial adenomatous polyposis (FAP) is an inherited autosomal dominant disease caused by a mutation in the APC (adenomatous polyposis coli) gene and/or a preneoplastic lesion (1,2). This is the best known and most common form of polyposis, with a prevalence of approximately 1 case per 10 000-20 000 people (1). Its onset occurs usually at an early age and carries a risk of colorectal cancer close to 100% if the patient does not receive timely treatment (1,3). The average age of cancer onset is 39 years, while the average age of death in patients who are not treated early is 42 years (4).
Total colectomy is the only effective treatment to prevent colon cancer in patients with FAP. Prophylactic colectomy should be done before the age of 25 years if the patients’ relatives have the APC gene mutation and polyps, as this may improve their life expectancy (4). The following is the case of two adolescent siblings with FAP.
Case 1
A 13-year-old female patient (older sister) was taken to the emergency room of a quaternary care institution in Bogotá (Colombia) in 2015 due to a 3-year history of bloody, glistening stools that worsened 3 days prior to her admission. She also presented with asthenia and adynamia.
The patient was hospitalized and required multiple transfusions of red blood cell units due to anemia with hemodynamic repercussions. Once she was stabilized, an upper gastrointestinal endoscopy and a total colonoscopy were performed 24 hours after admission, revealing chronic nodular gastritis and polyps in all segments of the colon (more than 100 in each segment, with a higher concentration in the transverse colon and rectum), respectively. In view of these findings, she underwent follow-up colonoscopies and polypectomies on the 1st, 23rd and 51st days of her hospital stay, during which samples were taken and sent to pathology for study. The pathology report showed multiple juvenile polyps, some of them hyperplastic and one of them with low-grade dysplasia (with lesion-free resection edges).
Based on the findings, the patient was diagnosed with FAP and, after 71 days of hospital stay, underwent a laparoscopic colectomy with transanal pull-through and ileostomy. During this procedure, it was noted that the colon was enlarged, had indurated and fibrotic walls, and was filled with polyps (above the pectineal line) larger than 1cm (Figures 1 and 2); the terminal ileum was healthy and had no polyps. At the end of this procedure, a protective ileostomy was left in place and the stoma was subsequently closed.
The patient, who had an adequate postoperative progress and was discharged after 80 days of hospital stay, remained asymptomatic in the routine outpatient follow-up.
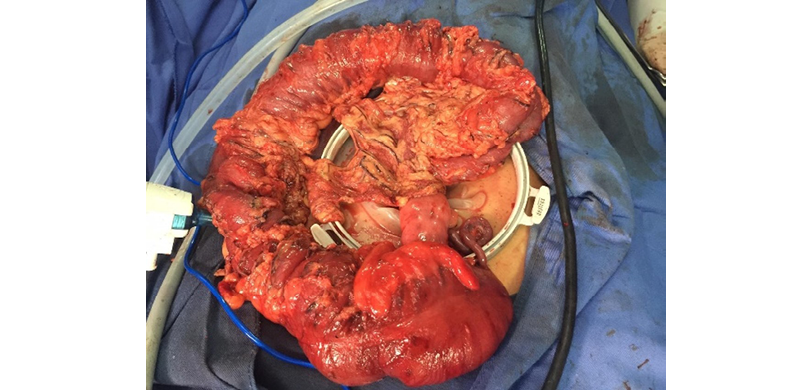
Figure 1. Surgical specimen showing familial adenomatous polyposis.
Source: Image obtained while conducting the study.
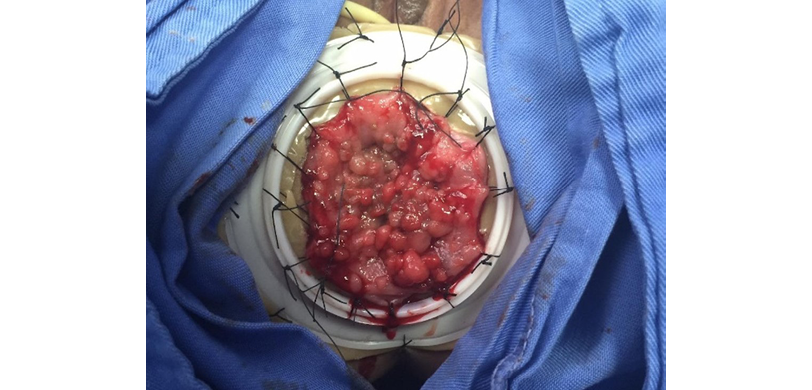
Figure 2. Multiple polyps detected during surgical removal.
Source: Image obtained while conducting the study.
Case 2
A 14-year-old male patient (younger brother) was taken to the emergency room of a quaternary care institution in Bogotá in 2021 due to an 8-month history of abundant bleeding during bowel movements. This patient had been diagnosed with FAP three years earlier by means of an anatomopathological examination performed on samples of lesions (polyps) obtained during a colonoscopy. After being diagnosed, the patient did not continue to be followed up.
On admission, an abdominal X-ray and complete blood count were requested in order to perform an elective total colectomy, taking into account the history described above. On the third day of hospitalization, total proctocolectomy with ileoanal anastomosis (J-pouch) and Hartmann’s procedure were performed. One week after the procedure, anoscopy with ileocolonoscopy was performed, which showed 40% dehiscence of the anastomosis with no communication with the abdominal cavity, so no additional procedures were performed.
When implementing the postoperative dilatation plan, there was resistance to the passage of the dilator, indicating stenosis of the anastomosis, which was managed with Hegar dilators; once an adequate caliber was achieved, the intestinal stoma was closed.
The patient had an adequate medical progress, so he was discharged 25 days after admission and continued to receive outpatient follow-up by the pediatric surgery service, without the need for readmission or further interventions. The histopathologic report revealed multiple juvenile polyps, some hyperplastic and others with low-grade dysplasia, and lesion-free resection edges (Figure 3).
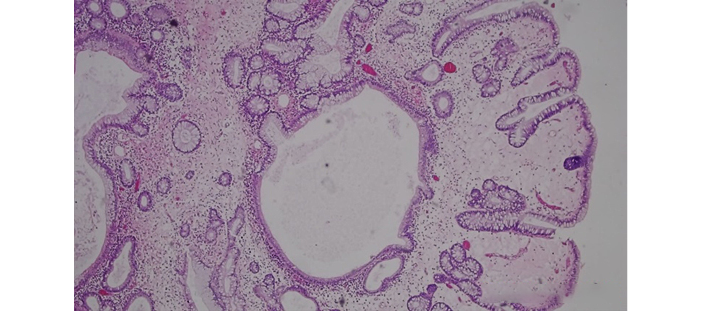
Figure 3. Histopathologic specimen. Juvenile polyps with glands of different shape and size, dilated, ulcerated and surrounded by abundant inflammatory stroma.
Source: Image obtained while conducting the study.
These two cases have the following family history: their mother died at 36 years of age from rectal cancer with K-RAS gene mutation confirmed by immunohistochemistry; their maternal grandmother, maternal aunt and maternal uncle died of colorectal cancer at 49, 51 and 22 years of age, respectively; a 34-year-old maternal uncle had a total colectomy for FAP; and two male and female maternal cousins, aged 32 and 37 years, respectively, were also diagnosed with FAP. In total, 7 first- and second-degree relatives had a history of FAP and/or colorectal cancer (Figure 4).
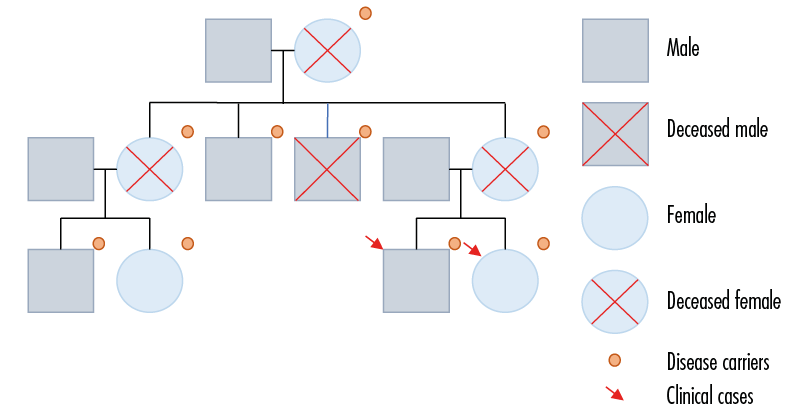
Figure 4. Genogram.
Source: Own elaboration.
Discussion
FAP is a preneoplastic syndrome characterized by the presence of hundreds of adenomatous polyps in the colon and upper digestive tracts. This disease is inherited in an autosomal dominant manner and is 100% penetrant, so every child of a patient with FAP has a 50% risk of suffering from it. It is caused by a mutation in the APC gene, which is located on chromosome 5q21 and plays an important role in the regulation of the intracellular levels of β-catenin (4). The accumulation of β-catenin causes loss of cell adhesion; transcription of homeotic genes involved in tissue polarity and architecture; and activation of cell proliferation, leading to uncontrolled growth of epithelial cells which, in turn, results in polyps (4,5). Spontaneous mutation accounts for 20% of FAP cases (1,4-6).
FAP is characterized by the presence of more than 100 polyps in the colon and rectum (1). The most common phenotypic expression of this disease is colorectal polyposis, but it can also affect other segments of the gastrointestinal tract and have extracolonic manifestations in up to 70% of cases, including desmoid tumors, hypertrophy of the retinal pigment epithelium, and lipomas (6).
The diagnosis of FAP is confirmed with the histopathological report of polypoid lesions observed in endoscopic studies requested to analyze gastrointestinal bleeding or by screening patients with a family history of FAP. It is ideal to perform a genetic study of the mutation in all patients at risk (7).
Prophylactic surgery is the most recommended treatment in patients diagnosed with FAP. Moreover, in these patients total colectomy is the only effective treatment to prevent colon cancer, and the surgical technique selected depends mainly on the severity of the clinical manifestation of the disease, the age of the patient at the time of diagnosis, and particular clinical considerations (1).
According to Cruz-Cortés et al. (6), in 1947 Ravitch and Sabiston described alternative techniques for patients requiring colectomy (including patients with FAP), among which complete proctectomy including the mucosa with ileoanal anastomosis and sphincter mechanism preservation and fecal continence are described. The basis of some of these techniques can be traced back to the pull-through described by Soave in 1965 for the surgical management of Hirschsprüng’s disease, in which the limits of the pectineal line are preserved. Cruz-Cortés et al. (6) also indicated that multiple treatment alternatives were modified by Kock, such as permanent colectomy and ileostomy or proctocolectomy with continent ileal pouch (6-8). The technique used in the cases reported here was total proctocolectomy + conversion to laparoscopic-assisted abdomino-perineal pull-through + ileoanal anastomosis with J-pouch + Hartmann’s ileostomy, which is currently the most widely accepted.
Total proctocolectomy with definitive ileostomy is indicated for patients with rectal cancer without need for reconstruction (5), while mucotomy is indicated in cases with diffuse involvement of the rectum, even up to the pectineal line. It should be noted that, although only case series have been found in this regard, it has been shown that prophylactic colectomy has increased the half-life of patients with FAP, which is why it is recommended (1,8).
The use of chemopreventive agents and non-steroidal anti-inflammatory drugs in patients with FAP is controversial. However, the latter have shown promising outcomes in some studies, especially sulindac and celecoxib (1), but further studies are needed to demonstrate the convenience of using this group of drugs, taking into account their possible adverse effects (1,3).
Screening of first-degree relatives of a patient with FAP should begin with lower endoscopy between the ages of 10 and 15 years. Also, sigmoidoscopy should be performed annually until the age of 25, then every two years until the age of 35, and then every 3 years until the age of 50. Patients with FAP who do not want to be treated surgically should undergo lower GI endoscopy every year, while those with total colectomy and rectal preservation should continue to be followed every 6-12 months because of the risk of developing rectal cancer. If adenomas are found, they should be resected promptly. Patients who underwent proctectomy with ileoanal pouch should continue to be followed up once a year due to the described cases of remnant adenomas in the rectum and the pouch, especially if mucotomy was not performed (1).
While there is no consensus on the follow-up of patients with FAP and extracolonic manifestations, if suspected, it has been proposed to perform an upper gastrointestinal endoscopy every 3-5 years, a CT scan of the abdomen every year in search of desmoid tumors, and a retinoscopy every 2-3 years (1); likewise, a periodic physical examination should be performed. In this respect, no studies that provide follow-up of these patients are available in order to clarify thoroughly the progression of these associated lesions.
A multidisciplinary approach is key to the management of patients with FAP and their relatives, in order to understand the disease and its natural history, as well as to know the available treatment options, understand the inheritance of this disease, and provide genetic counseling to establish the likelihood of its occurrence in offspring and other relatives. It is also important that patients receive psychological support to cope with disease and bereavement. All these strategies facilitate early diagnosis, reduce the morbidity and mortality of the family group, and help to accept the situation.
Conclusions
FAP is a rare inherited autosomal dominant disease in the pediatric population, which must be diagnosed in a timely manner to prevent the development of cancer. A comprehensive approach to this disease has an impact on patient morbidity and mortality. Surgical planning and approach are the mainstay for symptom control and cancer prevention; they must be adapted to each patient. Genetic counseling is fundamental for patients and their relatives, as it helps in making short-, medium- and long-term decisions.
Ethical considerations
The patients and their legal guardians signed the informed consent form, and this study was submitted for approval to the Ethics Committee of the Fundación Hospital Pediátrico de La Misericordia (HOMI).
Conflicts of interest
None stated by the authors.
Funding
None stated by the authors.
Acknowledgments
None stated by the authors.
References
1.Parés D, Pera M, González S, Pascual-Cruz M, Blanco I. Poliposis adenomatosa familiar. Gastroenterol Hepatol. 2006;29(10):625-35. https://doi.org/frs366.
2.Mougenot JF, Olshwang S, Peuchmar M. Intestinal Tumors: Intestinal Polyps and Polyposis. In: Walker WA, editor. Pediatric Gastrointestinal Disease: Pathology, Diagnosis, Management, Volume 1. 4th ed. BC Decker Inc.; 2004. p.214-223.
3.Latt TT, Nicholl R, Domizio P, Walker-Smith JA, Williams CB. Rectal bleeding and polyps. Arch Dis Child. 1993;69(1):144-7. https://doi.org/dfvtpd.
4.Chanis RA, McCalla R, Sánchez A, Rueda K, Ah Chu MS, Dutari JE, et al. Poliposis Adenomatosa familiar con transformación maligna en niños: presentación de casos. Pediátr Panamá. 2014;43(1):31-5.
5.Nacif PA, Caballero G, Gutiérrez C, Méndez V. Poliposis adenomatosa familiar. Presentación de dos casos. Arch Pediatr Urug. 2006;77(3):262-266.
6.Cruz-Cortés S, Mora-Fol JR, Ruelas-Vargas C, Chávez-Barrera JA. Evolución en el manejo quirúrgico de la Poliposis Adenomatosa Familiar con el uso de sutura mecánica en Pediatría. Rev Mex Cir Pediátr. 2009;16(4):156-61.
7.Cázares-Méndez JM, Zamudio-Vázquez VP, Gómez-Morales E, Ortiz-Aguirre SG, Cadena-León JF, Toro-Monjaraz EM, et al. Pólipos gastrointestinales en pediatría. Acta Pediatr Mex. 2015;36:158-63. https://doi.org/mzpf.
8.Velasco RA, Núñez RN, Hurtado CM, Zarabozo EE, García RC, Jaén CJ. Poliposis adenomatosa familiar: seguimiento de las manifestaciones extracolónicas. Cir Pediatr. 2010;23(1):35-9.