TWO FAMILIES WITH MIDD AND MELAS: HETEROPLASMY LEVEL IN m.3243A>G MUTATION AND THE FIRST REPORT ON THE m.3271T>C MUTATION IN COLOMBIA
Palabras clave: MELAS; Heteroplasmia; Enfermedad mitocondrial.
Keywords: MELAS; Heteroplasmy; Mitochondrial Disease.
Jorge L Granadillo
Physician, Master in Human Genetics
Universidad Nacional de Colombia
– Bogota Campus – Institute of Genetics
Bogota, D.C. – Colombia
Leonardo F Hernández–Reina
Neurologist
Fundación Universitaria de Ciencias de la Salud
– Hospital San José –
Departament of Neurology
Bogota, D.C. – Colombia
Juan Manuel Arteaga–Diaz
Endocrinologist
Universidad Nacional de Colombia
– Bogota Campus –
Department of Internal Medicine
Endocrinology Unit
Bogota, D.C. – Colombia
Manuel Luna
Neuropediatrician, Master in Bioethics
Hospital El Tunal
– Departament of Pediatrics –
Bogota, D.C. – Colombia
Clara E. Arteaga–Díaz
Physician, Magister in Human Genetics
Universidad Nacional de Colombia
– Bogota Campus –
Instituto de Genética
Bogota, D.C. – Colombia
Correspondencia:
Clara E. Arteaga–Díaz
Institute of Genetics,
Universidad Nacional de Colombia
– Bogota Campus –
Bogota, D.C. – Colombia.
Email: cearteagad@unal.edu.co
ABSTRACT
MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) and MIDD syndrome (maternally inherited diabetes and deafness) are mitochondrial diseases caused in most cases by the same mutation m.3243A> G, which affects the gene MT-TL1.
The cases of two families with MELAS are presented here. In the first case, the m.3243A>G mutation was detected and the heteroplasmy level in blood, urine and oral mucosa were determined, finding a great phenotypic variability: the patient had higher heteroplasmy in the three tissues compared against oligosymptomatic relatives, and the mother had high blood sugar levels and hearing loss, suggesting a phenotype near to MIDD. In the second family, the m.3271T>C mutation was detected, which constitutes the first case reported in Colombia.
These findings suggest that MIDD and MELAS, often described as distinct entities, are part of the same entity with variable expressivity partially depending on heteroplasmy.
Rev Case Rep. 2016; 2(1): 27–36
INTRODUCTION
MELAS is a multisystem disease characterized by the presence of mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes. Usually, the initial psychomotor development is normal and the condition typically starts developing during childhood. Other common clinical manifestations include ataxia, myoclonus, episodic coma, cardiomyopathy, pigmentary retinopathy, ophthalmoplegia, diabetes, hirsutism, gastrointestinal dysmotility, migraines and nephropathy.
This disease is caused by mutations in mitochondrial DNA (mtDNA) and the most frequent is m.3243A>G, which affects one of the genes of the mitochondrial leucine tRNA (MT-TL1) (1.2). The clinical manifestations of such mutation are highly heterogeneous and produce different clinical presentations, including MELAS, diabetes, maternally inherited deafness (MIDD) and even oligosymptomatic and asymptomatic individuals. The causes of this variability are not completely understood yet, but genetic or environmental factors may be involved (3-6).
Given that the mutation responsible for these diseases is found in the mitochondrial genome, that each cell has multiple mitochondria and that each mitochondrion has multiple copies of its DNA, it is common that such mutation is not present in all of them and that the frequency of this event varies from cell to cell, from tissue to tissue and from patient to patient. This phenomenon is known as heteroplasmy and is one of the genetic factors that has been considered as the responsible for the clinical variability (7).
Some studies have been conducted to evaluate the association between the heteroplasmy level in various tissues and the clinical manifestations in patients with MELAS and their relatives, finding higher levels of heteroplasmy in skeletal muscle cells, followed by urinary sediment cells. Higher levels of heteroplasmy are related to the presence of certain clinical features such as myopathy, seizures, stroke - like episodes, cardiomyopathy, short stature and low weight (5).
There is only one previous study in Colombia, in which two families with MELAS and m.3243A>G mutation were evaluated; the heteroplasmy behavior in these families and its correlation to clinical manifestations were analyzed (8) .
Two cases evaluated at the Institute of Genetics from Universidad Nacional de Colombia at its Bogota Campus are presented here. The first case deals with a family with MELAS and m.3243A>G mutation, to whom the level of heteroplasmy was evaluated in three tissues: blood, urine sediment and oral mucosa; then an analysis of the results and their relationship with the clinical picture of the individuals is made. The second case is about another family with MELAS and m.3271T>C mutation, which constitutes the first family case reported in Colombia with this mutation. Both families signed an informed consent during their first visit to the institute.
PATIENT INFORMATION
Family MELAS 1
The patient was a six-year old girl when first consulted, who started to develop unsteady gait and short stature at about three years of age, but had normal fine motor skills and language. At the age of five, she suffered the first convulsive episode, characterized by loss of consciousness, cyanosis, clonic movements of the head and sudden loss of tone in the rest of the body. Six months later, she presented the first stroke-like episode, characterized by seizures associated with vision loss. Two months later, she presented a similar episode and since then, has continued to show partial loss of vision, left hemiparesis, headache and recurrent vomiting. She is currently being treated with levetiracetam, oxcarbazepine, carnitine and clonazepam.
Important background information showed that the patient is the third daughter of a 30-year-old mother, non-consanguineous parents, pregnancy complicated only by gestational diabetes and full term delivery via C-section.
The patient had neonatal respiratory distress, but did not require intubation nor in-patient stay in the neonatal intensive care unit, and had apparently normal early neurodevelopment. Relevant aspects of family history are (Figure 1): mother with gestational diabetes during pregnancy; maternal half-sister and possibly the mother with migraine; maternal uncle with epilepsy since age 11, who apparently died of cerebrovascular disease at age 12; maternal second cousin with hemiparesis since birth; maternal grandmother with a history of two years of unspecific psychiatric disorder characterized by aggression and psychosis already resolved; no family history of diabetes mellitus, heart disease, chronic renal failure or deafness.

Fig 1. MELAS 1 family tree.
Source: Own elaboration based on the data obtained in the study
Physical examination found a thin (BMI = 12), normocephalic patient, with sluggishly reactive isochoric pupils, preserved eye movements, generalized hypotonia with left hemiparesis, poor coordination, predominant truncal ataxia and patellar reflexes +++/++++. Paraclinical studies were notable due to high lactate (2.24 mmol/L); creatinine, blood urea nitrogen and fasting blood sugar within normal ranges. Brain magnetic resonance imaging performed during the first stroke-like episode revealed a right temporo- occipital cerebral infarction, with spectroscopy that reported decreased peaks of N-acetylaspartate, choline and creatine, and a dominant peak of lactate in the right temporo- occipital area. The panel for prothrombotic conditions (Factor VIII, factor C, protein S, antithrombin III, protein C resistance, IgG and IgM) was within normal ranges.
The mother was 36 years old at the time of the first consultation and had a normal physical examination with normal body mass index (BMI). The maternal half-sister was 18 at the time of the first consultation and also presented normal physical examination and BMI.
The presence and level of heteroplasmy of m.3243A>G mutation was determined through the amplification refractory mutation system with quantitative PCR (ARMS-qPCR) as described by Wang et al. (9). The evaluated samples included urine sediment, oral mucosa smear and peripheral blood; the results of these tests are shown in Table 1. Additionally, other complementary tests were performed (Table 2).
Table 1. ARMS-qPCR results for m.3243A> G mutation in MELAS 1 family.
|
Individual |
Sample |
Heteroplasmy (%) |
Interpretation |
|
Patient |
Blood |
80 |
+ |
|
Urine |
97 |
+ |
|
|
Oral mucosa |
90 |
+ |
|
|
Mother |
Blood |
16 |
+ |
|
Urine |
68 |
+ |
|
|
Oral mucosa |
15 |
+ |
|
|
Sister |
Blood |
47 |
+ |
|
Urine |
40 |
+ |
|
|
Oral mucosa |
57 |
+ |
Source: Own elaboration based on the date obtained in the study.
Table 2. Complementary tests for MELAS 1 family.
|
Individual |
Fasting Glucose (mg/dL) |
Blood creatinine (mg/dL) |
Urea Nitrogen (mg/dL) |
Glomerular filtration rate (mL/ min/1.7 3 m2) |
Tonal audiometry |
EKG |
|
Patient |
81 |
0.331 |
24.9 |
143.5 |
Normal |
Normal |
|
Mother |
108 |
1.11 |
17.1 |
63 |
Mild bilateral hearing loss |
Normal |
|
Sister |
88 |
0.91 |
19.6 |
92 |
Normal |
Right bundle branch block |
Source: Own elaboration based on the data obtained in the study.
Family MELAS 2
The patient was a 24-year-old man at the time of the first consultation, who started to develop multiple stroke-like episodes associated with focal epilepsy at age 18; the first time, he presented myoclonic movements of the left upper limb. In addition, the patient had a history of recurrent migraines -which had required in-patient hospital care-, decreased visual acuity, recurrent vomiting associated with hiatal hernia, gastroesophageal reflux disease and Helicobacter pylori infection; he is currently being treated with clonazepam, levetiracetam, coenzyme Q10 and L-carnitine.
Important background information reported that the patient is the second child of a 34-year-old mother with non-consanguineous parents, pregnancy and childbirth without complications and with normal initial neurodevelopment. Family history is notable due to the presence of maternal relatives with diabetes mellitus, migraine and deafness (Figure 2). The patient’s mother had a history of varicose disease in lower limbs and gastric carcinoma at 50 years of age.
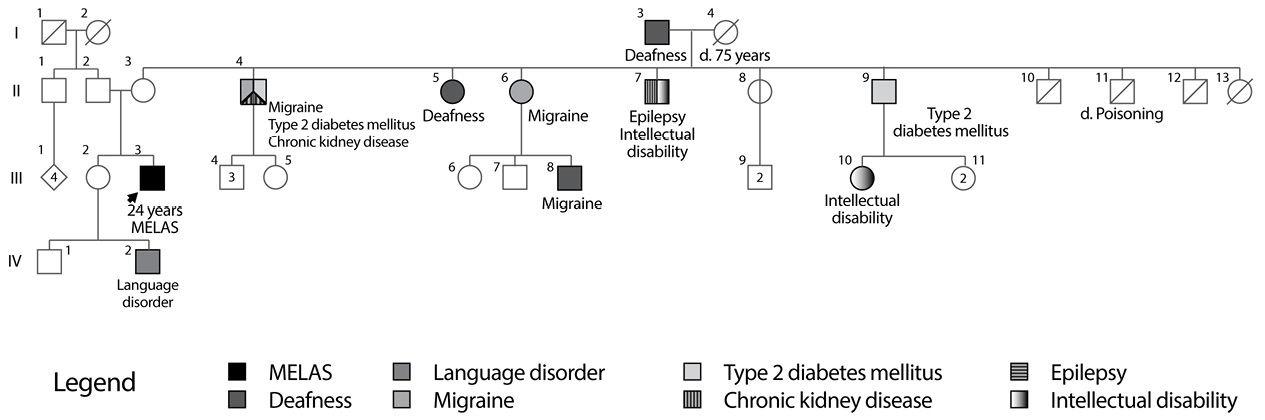
Fig 2. Family Tree MELAS 2 Family.
Source: Own elaboration based on the data obtained in the study
Physical examination found a thin patient, alert and oriented, but with mild bradypsychia and affect tending to flat. The rest of the neurological examination was normal, with no remarkable motor or sensory deficits. Laboratory and imaging studies showed normal fasting glucose, creatinine and blood urea nitrogen; transesophageal echocardiography without evidence of heart disease; brain MRI with multiple strokes, basal and medial right occipito- temporal predominance; multiple abnormal electroencephalograms that reported slow activity in the right hemisphere, and generalized polyspike and slow waves; elevated lactic acid in blood before and after physical activity and auditory evoked potential compatible with mild bilateral peripheral sensorineural hearing loss. The patient underwent a mitochondrial mutation panel in peripheral blood through restriction fragment length polymorphism (RFLP), reporting the presence of m.3271T>C mutation associated with MELAS and discarding mutation m.3243A>G, among others.
DISCUSSION
This paper presents the case of two patients with MELAS associated with two different mutations, both consisting of transitions subjected to the phenomenon of heteroplasmy and found in the MT-TL1 gene, which is part of the mitochondrial DNA.
First, a family with m.3243A>G mutation, whose family tree shows the already known phenotypic heterogeneity of this mutation, as well as individuals with mild manifestations (mother and half-sister), and others with MELAS with a more severe picture (the patient and probably her maternal uncle) is reported. Although the sample size is small to determine whether there is a correlation between the level of heteroplasmy and the clinical manifestations, it is evident that, in this case, the patient has a heteroplasmy level in the three tissues analyzed, much higher than her slightly affected relatives, which supports the hypothesis that the heteroplasmy level is related to the number and severity of clinical manifestations. Both the mother and the patient showed higher heteroplasmy levels in the urinary sediment and lower levels in blood, which was not the case of her half - sister, in which the most affected tissue was the oral mucosa, followed by blood and urine sediment.
Previous studies show that urinary sediment usually reveals higher heteroplasmy, followed by the oral mucosa and peripheral blood. The cause is not entirely known, but this probably happens due to two factors: 1) the rate of cell division and 2) tissue mutational threshold. For example, tissues with increased energy expenditure show alterations with smaller mutational loads. Peripheral leukocytes have a high energy expenditure and also divide rapidly, so those with a high mutational load would be particularly sensitive to secondary mitochondrial dysfunction and, thus, would be subjected to a strong negative selection, which would result in lower heteroplasmy with time; on the other hand, the urinary sediment consists of transitional cells with rapid cell division but with lower energy requirements, giving them greater tolerance to mitochondrial dysfunction and allowing them to increase their mutational load (5,6).
Other studies have addressed the issue of the existence of a threshold effect in diseases secondary to mitochondrial mutations; that is, a heteroplasmy level at which clinical manifestations develop. It has been proposed that this phenomenon occurs because mitochondria under normal conditions have more mRNAs, tRNAs and active respiratory chains that are needed for normal cellular respiration, which allow tissues to tolerate a load of deleterious mitochondrial mutations up to a certain percentage, which constitutes the threshold (10). The best example is the m.8993T>G mutation, in which individuals with less than 60% heteroplasmy —in muscle— are usually asymptomatic or have only mild pigmentary retinopathy or migraines. Individuals with heteroplasmy ranging between 70% and 90% develop NARP syndrome (Neuropathy, ataxia, and retinitis pigmentosa) and individuals with heteroplasmy higher than 90% develop Leigh’s disease, a neurodegenerative and fatal disorder (11).
In the case of MELAS, MIDD and the m.3243A>G mutation, the correlation is not clear and there is no precise threshold; however, several studies have found a phenotypic threshold for MELAS ranging between 60% and 90% heteroplasmy in muscle (10), which reveals a large overlap between asymptomatic individuals and oligosymptomatic individuals with MIDD and MELAS. No studies attempting to determine the value of the phenotypic threshold of this mutation in other tissues have been found.
On the other hand, it is important to note that the phenotypic manifestations of the mutation in question are not static and may change over time. In this vein, it is worth analyzing the case of the mother of the girl, whose physical examination was completely normal but presented altered fasting glucose and mild hearing loss; the progression of these disorders could lead to diabetes and maternally inherited deafness, a clinical picture that makes part of the clinical spectrum of this mutation. In fact, this individual had gestational diabetes during the pregnancy of the patient, situation previously reported by Laloi-Michelin et al., for a group of MIDD patients, of which about 16% had gestational diabetes as initial presentation (12).
Although the m.3243A>G mutation does not seem to be a common cause of gestational diabetes (13), this finding shows that mitochondrial dysfunction may play a role in the pathogenesis of this type of diabetes, since a recent study shows a reduction in mitochondrial protein expression and altered calcium signaling proteins in the skeletal muscle of women with gestational diabetes (14).
On the other hand, a more detailed analysis of this patient’s audiometry reveals that her hearing loss mostly affects the highest frequencies (6000-8000Hz), which is a common feature of deafness secondary to this mutation, initially affecting this type of frequencies and subsequently progressing to all frequencies. In this regard, MIDD-related hearing loss has a similar course as presbycusis, except that the latter starts at a much later age; nevertheless, hearing loss in patients with MIDD usually appears after diabetes (15), which is not the case of this patient and corroborates that deafness is not secondary to chronic hyperglycemia, but probably to the underlying mitochondrial dysfunction. These findings may suggest that MIDD and MELAS, often described as distinct entities, are part of the same entity with variable expressivity, depending in part on heteroplasmy.
Another important finding was the presence of a right bundle branch block in the half-sister of the patient, whose physical examination was also normal. Since the myocardial tissue is a high energy consumer and depends on the B-oxidation of fatty acids MIDD —which occurs in the mitochondrae — as an energy source, it is not surprising that the heart is often affected in mitochondrial diseases. Even when the most common involvement is usually cardiomyopathy or Wolf-Parkinson-White syndrome (WPW), a report of Hinaro et al. reported a prevalence of 6% of cardiac conduction blocks in patients with MELAS (16) and a Japanese study revealed a prevalence of 10% of cardiac conduction disorders in diabetic patients with m.3243A>G mutation (17). Similarly, a Dutch study obtained a prevalence of 25% of electrocardiographic abnormalities in asymptomatic individuals who carry this mitochondrial mutation (6).
This highlights the importance of performing a molecular diagnosis of the maternal relatives of affected individuals, as well as a close clinical monitoring that examines those who exhibit the mutation, even if their symptoms are mild or nonexistent. There is no consensus about the management of asymptomatic or oligosymptomatic maternal relatives, but a periodical evaluation by audiology and ophthalmology, and complementary testing (such as electrocardiogram, echocardiogram, blood glucose, blood insulin, fasting C-peptide and hemoglobin A1C) is considered appropriate in order to detect these diseases early and make an early intervention.
Second, the case of the MELAS 2 family with m.3271T>C mutation, which is the second most common cause and accounts for about 7.5% of MELAS cases (2), was presented; it has also been reported as the cause of MIDD (14,18). As far as known, this is the first case of this mutation reported in Colombia.
For this family, the molecular test was not performed to maternal relatives nor the heteroplasmy level was determined because the used methodology did not provide that information; however, the family tree reported multiple individuals with diabetes mellitus, deafness and migraine, manifestations likely to be related to the mutation, which also reflect the phenotypic heterogeneity of mutations in the MT-TL1 gene.
The case of the individual II-4, which has type 2 diabetes, migraine and chronic renal failure, is of interest. There are reports of individuals with m.3243A>G mutation and kidney diseases, including focal segmental glomerulosclerosis and tubulointerstitial nephropathy (1); as a matter of fact, individuals with MIDD have a higher risk of diabetic nephropathy compared to those with common diabetes mellitus, as previously explained, probably because cells of renal tubules have a high energy consumption (19). It is important, for future studies, to determine the presence and level of heteroplasmy in different members of this family, because, so far, no studies that evaluate the relationship between the level of heteroplasmy of m.3271T>C mutation and its clinical manifestations have been found. In addition, it is worth noting that both patients had cerebral ischaemia implicating posterior regions of the cerebral cortex. This is typical in MELAS and the reason for this distribution is currently unknown.
Betts et al. conducted a molecular and neuropathological study in two individuals with MELAS, which found evidence of mitochondrial dysfunction in the blood vessels of leptomeninges and the cerebral cortex, but this dysfunction was not limited to a specific brain region. In this work, the authors proposed that episodes of cerebral ischemia are related not only to mitochondrial dysfunction of blood vessels, but also to a phenomenon related to migraine, which is common in MELAS patients, known as cortical spreading depression (CSD).
The CSD is a cortical depolarization wave originated in the occipital region and then propagates anteriorly over the cerebral cortex, immediately followed by a period of inhibition of electrical activity. This phenomenon is accompanied by changes in the cortical vascular flow, initially presenting hyperaemia of approximately three to four minutes, followed by a slight hypoperfusion for about one to two hours. This hypoperfusion, together with the underlying mitochondrial dysfunction, would predispose cerebral ischaemia in posterior regions of the cerebral cortex (20).
In conclusion, the cases presented here confirm the great clinical variability of mutations of the MT-TL1 gene found in these families, especially in the case of m.3243A>G mutation in MELAS 1 family, whose heteroplasmy could explain the broad phenotypic spectrum within which MELAS and MIDD are included.
FUNDING
This study was funded by “Convocatoria para el estimulo a la investigación a través de proyectos y trabajos de investigación en los Posgrados de la Facultad de Medicina de la Universidad Nacional de Colombia – Apoyo a la Investigación en Salud – 2012”.
CONFLICTS OF INTEREST
None stated by the authors.
REFERENCIAS
1.Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. http:// doi.org/ffwh5f.
2.DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallac SE, Amemiya A, Bean LJH, et al. editors. GeneReviews. Seattle: University of Washington; 2001 [updated 2013 Nov 21; cited 2016 AprApr 28]. Available from: http://goo.gl/DWVjAQ..
3.van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1992;1(5):368-71. http://doi.org/fnhkqx.
4.Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann. N. Y. Acad. Sci. 2008;1142:133- 58. http://doi.org/c584zj.
5.Ma Y, Fang F, Yang Y, Zou L, Zhang Y, Wang S, et al. The study of mitochondrial A3243G mutation in different samples. Mitochondrion. 2009;9(2):139-43. http://doi.org/b5rtg7.
6.de Laat P, Koene S, van den Heuvel LP, Rodenburg RJ, Janssen MC, Smeitink JA. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A > G mutation. J. Inherit. Metab. Dis. 2012;35(6):1059-69. http://doi.org/bjn8.
7.Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann. Med. 2005;37(3):222-32. http://doi.org/cc6dfx.
8.Parra-Marin MV, Cornejo-Ochoa JW, DuqueéVélez CE, Ruiz-Linares A, Bedoya-Berrio G. Comportamiento de la mutación mtDNA A3243G en dos familias antioqueñas de pacientes diagnosticados con el síndrome MELAS. Iatreia. 2010;23(1):21-33.
9. Wang J, Venegas V, Li F, Wong LJ. Analysis of Mitochondrial DNA Point Mutation Heteroplasmy by ARMS Quantitative PCR. In: Current Protocols in Human Genetics. New york: John Wiley & Sons Inc.; 2001.
10.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370(3):751-62. http://doi.org/c9tnsb.
11.Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Karppa M, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am. J. Hum. Genet. 1998;63(2):447-54. http://doi.org/b7tp8g.
12.Guillausseau PJ, Dubois-Laforgue D, Massin P, Laloi-Michelin M, éBellanné-Chantelot C, Gin H, et al. Heterogeneity of diabetes phenotype in patients with 3243 bp mutation of mitochondrial DNA (Maternally Inherited Diabetes and Deafness or MIDD). Diabetes Metab. 2004;30(2):181-6. http://doi.org/cnm7kc.
13.Malecki M, Klupa T, Wanic K, Frey J, Cyganek K, Sieradzki J. Search for mitochondrial A3243G tRNA(Leu) mutation in Polish patients with type 2 diabetes mellitus. Med. Sci. Monit. 2001;7(2):246-50.
14.Murphy R, Turnbull DM, Walker M, Hattersley AT. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. 2008;25(4):383- 99. http://doi.org/dnpnjh.
15.Guillausseau PJ, Massin P, Dubois-LaForgue D, Timsit J, Virally M, Gin H, et al. Maternally inherited diabetes and deafness: a multicenter study. Ann. Intern. Med. 2001;134(9 Pt 1):721-8. http://doi.org/bjn9.
16.Katulanda P, Groves CJ, Barrett A, Sheriff R, Matthews DR, McCarthy MI, et al. Prevalence and clinical characteristics of maternally inherited diabetes and deafness caused by the mt3243A > G mutation in young adult diabetic subjects in Sri Lanka. Diabet. Med. 2008;25(3):370-4. http://doi.org/cngqtj.
17.Suzuki S, Oka Y, Kadowaki T, Kanatsuka A, Kuzuya T, Kobayashi M, et al. Clinical features of diabetes mellitus with the mitochondrial DNA 3243 (A-G) mutation in Japanese: maternal inheritance and mitochondria-related complications. Diabetes Res. Clin- Pract. 2003;59(3):207-17. http://doi.org/ccr7d8.
18.Tsukuda K, Suzuki Y, Kameoka K, Osawa N, Goto Y, Katagiri H, et al. Screening of patients with maternally transmitted diabetes for mitochondrial gene mutations in the tRNA[Leu(UUR)] region. Diabet Med. 1997;14(12):1032-7. http:// doi.org/b5bhs9.
19.Massin P, Dubois-Laforgue D, Meas T, Laloi-Michelin M, Gin H, Bauduceau B, et al. Retinal and renal complications in patients with a mutation of mitochondrial DNA at position 3,243 (maternally inherited diabetes and deafness). A case-control study. Diabetologia. 2008;51(9):1664-70. http://doi.org/cqtwb5.
20.Saker PJ, Hattersley AT, Barrow B, Hammersley MS, Horton V, Gillmer MD, et al. UKPDS 21: low prevalence of the mitochondrial transfer RNA gene (tRNA(Leu(UUR))) mutation at position 3243bp in UK Caucasian type 2 diabetic patients. Diabet. Med. 1997;14(1):42-5. http://doi.org/bmwg47.