RESUMEN
El leiomiosarcoma hepático primario es un tumor extremadamente raro entre los casos de tumores hepáticos en adultos, presentando una incidencia de 0.1 y 1%. En este artículo se presenta el caso de un hombre de 55 años con cuadro clínico de cinco meses de evolución que consiste en dolor abdominal, presencia de masa dura, no móvil, de gran tamaño, pérdida de peso, disnea y fiebre.
La tomografía axial computarizada (TAC) trifásica mostró masa hipercaptante en su periferia durante fase arterial hipodensa en su centro, localizada en segmentos VII y VIII, sin plano de separación del hígado. Debido a su sintomatología, el paciente fue intervenido quirúrgicamente por laparatomía exploratoria, hallándose masa cerebroide de 40 x 40cm, razón por la cual se practicó una tumorectomía sin hepatectomía, dejando bordes quirúrgicos libres.
El diagnóstico se realizó, en gran parte, mediante evaluación histopatológica, observándose estroma con formas multinucleadas pleomórficas, desmina (+), SMA (actina de musculo liso) y MSA (actina músculo específica) (+), Ki67 (+) y negativo para S100 (proteína S100) y anticuerpo CD117, lo que confirmó el diagnóstico de leiomiosarcoma pleomórfico de alto grado. Una vez presentó una notoria mejoría, el paciente fue dado de alta a los 16 días de su ingreso y fue derivado al servicio de oncología para su adyuvancia con quimioterapia.
Dado el tamaño de la masa encontrada, el pronóstico fue poco alentador, lo que hizo que el tratamiento quirúrgico fuera la única opción que ofreciera expectativas de supervivencia mediante hepatectomías regladas o “atípicas” con márgenes de seguridad e incluso trasplante hepático. Teniendo en cuenta lo anterior, se optó por la primera opción; tras seis meses de la cirugía, observándose mejoría clínica, además del tratamiento adyuvante, el paciente, aún con pronóstico desfavorable, permanecía estable, asistiendo a controles bajo manejo médico multidisciplinario.
Introducción
El leiomiosarcoma hepático primario (LHP) es un tumor extremadamente raro. Para 2011, se habían reportado en la literatura inglesa menos de 50 pacientes con este tipo de tumor (1,2). En el contexto de este estudio, es importante decir que, a la fecha, no existen reportes de casos similares en Ecuador y que, para 2014, eran muy pocos los casos reportados en América Latina (3).
En población adulta, los sarcomas hepáticos primarios constituyen un grupo de tumores excepcionales, representando entre 0.1 y 1% de todos los tumores hepáticos malignos existentes en esta población (4). Por lo general, estos se desarrollan en el útero, el retroperitoneo, los órganos genitales, los pulmones, en el hígado y en los grandes vasos (5); por su parte, el LHP surge a partir de las células musculares lisas de las estructuras vasculares intrahepáticas, de los conductos biliares o del ligamento redondo (6).
Su rareza, sus manifestaciones de imagen y su presentación clínica no específica dificultan un diagnóstico temprano (7). En la actualidad, su diagnóstico puede realizarse antes de operar, mediante citología o biopsia percutánea guiada por imagen, o después de operar (8-12). Sin embargo, algunas veces el diagnóstico diferencial entre sarcoma hepático primitivo o metastásico puede presentar dificultades (9), razón por la que el estudio anatomopatológico es fundamental. El diagnóstico histopatológico se caracteriza por la presencia de infiltrados difusos y uniformes en las células en forma de huso con núcleos hipercromáticos (8,9), mientras que en inmunohistoquímica se observa reacción positiva para desmina, SMA, MSA, Ki67 y negativo para S100 y CD117.
La cirugía resectiva es la mejor opción de tratamiento de esta condición; en esta se deben mantener los mismos principios quirúrgicos preconizados en la cirugía de sarcomas de partes blandas, siendo las resecciones hepáticas con márgenes oncológicos la elección más apropiada o el gold standard. No obstante, y debido al avanzado estado de la enfermedad al momento de su diagnóstico, la tumorectomía o enucleación seguida de tratamiento con quimioterapia coadyuvante puede ser otra opción terapéutica a tenerse en cuenta, incluso en casos con metástasis (10), lo que deja al trasplante hepático como última opción.
Caso clínicO
Paciente masculino de 55 años de edad, profesional con labores de oficina sin antecedentes médicos de importancia, quien niega consumir o haber consumido alcohol y tabaco y reporta antecedentes familiares de diabetes mellitus, cáncer de esófago y melanoma ocular. Presenta cuadro clínico de cinco meses de evolución caracterizado por dolor abdominal, de tipo opresivo y de moderada intensidad, localizado en hipocondrio derecho y mesogastrio, acompañado de masa abdominal dura, no móvil, de aproximadamente 20cm de diámetro, dolorosa a palpación (ver Figura 1), hiporexia y pérdida de peso, aproximadamente 45 libras en 100 días, disnea de medianos esfuerzos y fiebre vespertina de un mes de evolución previo a ingreso hospitalario. Con signos vitales estables.

Fig 1. Morfología del paciente. Masa tumoral de gran tamaño en hemiabdomen derecho.
Fuente: Imagen obtenida durante la realización del estudio.
A través de los exámenes de laboratorio se observa anemia leve normocítica hipocrómica Hb: 10,1mg/dl, pruebas de función hepática y colestásicas normales, tiempos de coagulación normales y reactantes de fase aguda sin alteraciones. También se obtienen resultados normales de marcadores tumorales como CAE, alfafetoproteína, CA 125, CA 15-3, CA 19-9 y CA 72-4. La ecografía de abdomen muestra masa tumoral en parte inferior del hígado de aproximadamente 30 x 20cm, hipoecoica y vascularizada (ver Figura 2). Por otra parte, la radiografía simple de abdomen permite observar imagen radiopaca en hipocondrio y flanco derechos. La angio-tomografía de abdomen trifásica muestra masa hipercaptante en su periferia, centro hipodenso en fase arterial heterogénea en fase portal y tardía, localizada en segmentos hepáticos V y VI, sin plano de separación del hígado más adenomegalias retroperitoneales (ver Figura 3).

Fig 2. Ecografía de abdomen 2D. Masa hepática de gran tamaño, hipoecogénica, con vascularización.
Fuente: Imagen obtenida durante la realización del estudio.
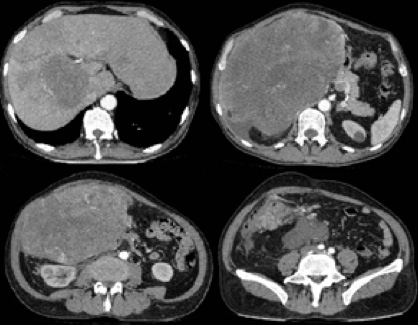
Fig 3. Angio-tomografía trifásica. Masa tumoral de gran tamaño en lóbulo hepático derecho, segmentos V y VI, que desplaza órganos intraabdominales.
Fuente: Imagen obtenida durante la realización del estudio.
Finalmente, mediante colonoscopía se observó mucosa de aspecto normal y disminución de luz a nivel de colon ascendente por aparente compresión extrínseca. Se practicó laparotomía exploratoria para mejorar el cuadro clínico del paciente, encontrando lo siguiente:: tumoración tipo cerebroide de gran tamaño en lecho hepático, segmentos V y VI, de aproximadamente 40x40cm, encapsulada, que desplazaba diafragma derecho y estructuras abdominales vecinas, por lo que se realizó tumorectomía sin hepatectomía, con bordes quirúrgicos libres en muestra por congelación; además, se liberaron estructuras vecinas y no se identificaron masas, ganglios o siembras metastásicas a simple vista. No se presentaron complicaciones quirúrgicas intraoperatorias (ver Figura 4).

Fig 4. Visualización, durante laparotomía exploratoria, de leiomiosarcoma hepático primario, multilobulado, tipo cerebroide, encapsulado, localizado en lóbulo hepático derecho.
Fuente: Imagen obtenida durante la realización del estudio.
El estudio anatomopatológico (ver Figura 5) reportó tumor maligno multilobulado con las siguientes características: peso, 4341 g; tamaño, 38 x 35 x 20cm; encapsulado, con bordes irregulares y cavitados; de estirpe mesenquimal compuesto por células fusiformes; con núcleo alargado; cromatina en grumos, y numerosas mitosis. Por su parte, el estudio inmunohistoquímico evidenció: desmina positivo, SMA y MSA positivos, Ki67 positivo en 80% de las células y negativo para S100 y CD117, lo que confirmó diagnóstico de leiomiosarcoma pleomórfico de alto grado.

Fig 5. Estudio histológico e inmunohistoquímico. 5.a. Se observan células malignas musculares fusiformes de núcleo alargado, con zonas de necrosis, las cuales son típicas en esta clase de tumor. (Tinción con hematoxilina-eosina x100). 5.b. Presencia de: numerosas mitosis, fascículos entrelazados y citoplasma rosado con numerosas formas multinucleadas pleomórficas. (Tinción con H&E x400).
5.c. Inmunohistoquímica muestra desmina positivo con presencia de células típicas de músculo liso. (Magnificación original: x50).
Fuente: Imagen obtenida durante la realización del estudio.
El paciente estuvo hospitalizado 16 días; luego de mostrar una evolución favorable, fue dado de alta y se ordenó control mensual por consulta externa. Asimismo, fue derivado al servicio de oncología para iniciar tratamiento adyuvante con quimioterapia cada 25 días por tres ciclos, observando su evolución clínica y ajustándose a su requerimiento con posibilidad de radioterapia posterior a control de parámetros clínicos, imagenológicos y de laboratorio, al cual se está a la espera en la actualidad. Luego de seis meses de postoperatorio, aún con pronóstico desfavorable, permanece estable, atendiendo a controles bajo manejo médico multidisciplinario.
DiscusióN
Las manifestaciones clínicas de LHP no son específicas y, por lo general, los tumores son asintomáticos antes de su aumento de tamaño (11,12). La edad promedio de presentación está entre los 40 y 50 años, con edades extremas de 22 y 77 años (13). El síntoma más frecuente es dolor, de pequeña a mediana intensidad, en hemiabdomen superior, acompañado de pérdida ponderal, febrícula vespertina y astenia (14). Mediante exploración clínica es posible identificar hepatomegalia o masa abdominal palpable en hipocondrio derecho-epigastrio. Por lo general, se describen elevaciones inespecíficas de algún parámetro bioquímico de función/daño hepático: bilirrubina, fosfatasa alcalina o transaminasas, mas no de marcadores tumorales específicos, como ocurrió en el caso aquí reportado, algo que sí si sucede en sarcomas presentes en otras partes del cuerpo.
Por su parte, el diagnóstico por imagen no aporta datos específicos (16,17), pues a través de ecografía se evidencia como una tumoración hepática hipoecoica, mientras que en TAC trifásica, como masa heterogénea bien definida, hipodensa o isodensa por áreas centrales de necrosis y con realce periférico, o como masa quística con pared gruesa (18) y con patrón angiográfico de masa avascular o importante neovascularización periférica patológica (19), superponible a cualquier tumor hepático. Finalmente, mediante resonancia magnética nuclear se observa un área heterogénea con lesión hipointensa en T1 e hiperintensa en T2 con posible encapsulación (2).
La localización más frecuente del tumor se da en lóbulo derecho y es común que a la hora de realizar el diagnóstico se encuentre metástasis en alrededor del 40% (6), lo que concuerda con los datos reportados aquí. Por tanto, el diagnóstico diferencial debe establecerse entre diversos tipos de tumores sólidos hepáticos benignos (20,21) y malignos, tales como hepatocarcinomas de diferente estirpe (22), sarcoma primitivo o metastásico (8) e incluso sarcomas de la vena cava retrohepática (23).
En la actualidad y En la mayoría de series de casos (20,24,25), el diagnóstico de sarcoma hepático puede establecerse preoperatoriamente, mediante citología o biopsia percutánea guiada por imagen; sin embargo, si la lesión hepática aparenta malignidad y se considera resecable su diagnóstico se realiza postoperatoriamente, tal como ocurrió en este caso. El diagnóstico histopatológico muestra cuatro tipos de leiomiosarcomas: 1, 2, 3 y 4, los cuales se definen como bien diferenciado, moderadamente diferenciado, pobremente diferenciado y leiomiosarcoma mixoide, respectivamente (26). En el presente caso el paciente fue clasificado en el tipo 1, pues el estudio inmunohistoquímico evidenció desmina y SMA Positivos, pero negativo para S-100, CD117 y NSE (18,19), lo que concuerda con los parámetros de esta tipología y confirmando así así el diagnóstico de LHP.
Al ser una masa hepática de gran tamaño que genera dificultad respiratoria por compresión a estructuras vecinas y que presenta una nutrida vascularización periférica, la posibilidad de toma percutánea guiada por imagen es descartada debido al alto riesgo de sangrado. Las hepatectomías regladas o atípicas con márgenes de seguridad son el tratamiento de elección, no obstante, y debido al avanzado estado evolutivo de la enfermedad al momento de su diagnóstico, la enucleación seguida de tratamiento quimioterapéutico es otra opción terapéutica, incluso en casos con metástasis (8), la cual, en el presente caso, fue a priori la mejor opción, siendo un caso excepcional con aparente ausencia de metástasis y bordes quirúrgicos libres.
Al ser este el único tratamiento que permite expectativas de sobrevivencia prolongada, su análisis revela los principales factores pronósticos favorables: edad inferior a 50 años, diagnóstico precoz con tamaño inferior a 5 cm, localización del tumor, cirugía radical con márgenes de seguridad, tratamiento adyuvante con quimioterapia y, como última opción, trasplante hepático (9). En un estudio, King et al. (27) describen casos con grandes tumores que luego de cinco años presentaban una tasa de supervivencia del 18%, así como casos con cerca de 80% tumores pequeños con márgenes libres. Por su parte, Gates et al. (28) señalan que la combinación de cirugía con quimioterapia ofrece una supervivencia media de 3.3 años.
Los LHP pueden presentar metástasis principalmente vía hematógena hacia el pulmón, seguido de vía linfática y peritoneal. Al respecto, Shivathirthan et al. (29) describen que el rango intermedio en la identificación de metástasis entre leiomiosarcoma primario y LHP fue de 29 meses (rango: 6-58 meses). Además, señalan que los criterios de inoperabilidad pueden incluir: propagación extrahepática del tumor, tumor intrahepático difuso y función hepática deteriorada (29).
El paciente del presente caso ha recibido 3 ciclos de quimioterapia con ifosfamida + doxorubicina; igualmente, ha asistido a controles mensuales clínicos, de laboratorio y de imagenología. Sin embargo, el rol de la adyuvancia con quimioterapia/radioterapia no está bien definido, pues a pesar de que la quimioterapia en forma de doxorubicina e ifosfamida muestra un curso lento de la enfermedad y puede prolongar la supervivencia en resecciones con estadio R1, n hay suficiente evidencia en tumores irresecables y en metástasis por LHP (30).
El rol del trasplante hepático aún es controversial, pues tiene porcentajes bajos de supervivencia y recurrencias del 95% antes de los seis meses (31).
ConclusioneS
El LHP es un tumor extremadamente raro que, en la mayoría de los casos, se diagnostica en estadios avanzados, lo que retrasa su tratamiento y empeora su pronóstico. Su hallazgo debe ser sospechado en pacientes con masas tumorales de gran tamaño. Ahora bien, a pesar de contar con gran cantidad de estudios avanzados de imagen, su diagnóstico es totalmente histopatológico, mientras que su tratamiento, quirúrgico, en la mayoría de los casos y dependiendo de varios factores. Este caso destaca la terapia quirúrgica y el diagnóstico de este tumor tan poco frecuente.
Financiación
Ninguna declarada por los autores.
Conflicto de intereses
Ninguno declarado por los autores.
Referencias
1.Ferrozzi F, Bova D, Zangrandi A, Garlaschi G. Primary liver leiomyosarcoma: CT appearance. Abdom Imaging. 1996;21(2):157-60. http://doi.org/cbnmww.
2.Soyer P, Blanc F, Vissuzaine C, Marmuse JP, Menu Y. Primary leiomyosarcoma of the liver MR findings. Clin Imaging. 1996;20(4):273-5. http://doi.org/d59c7d.
3.Del Carmen Binda M. Editorial de contenido. Rev Argent Radiol. 2014;78(1):3-4. http://doi.org/bt9z.
4.Liver Cancer Study Group of Japan. Primary liver cancer in Japan: Sixth report. Cancer. 1987;60(6):1400-11. http://doi.org/cvxkns.
5.Cioffi U, Quattrone P, De Simone M, Bonavina L, Segalin A, Masini T, et al. Primary multiple epithelioid leiomyosarcoma of the liver. Hepatogastroenterology. 1996;43(12):1603-5.
6.Civardi G, Cavanna L, Iovine E, Buscarini E, Vallisa D, Buscarini L. Diagnostic imaging of primary hepatic leiomyosarcoma: a case report. Ital J Gastroenterol. 1996;28(2):98–101.
7.Chi M, Dudek AZ, Wind KP. Primary hepatic leiomyosarcoma in adults: analysis of prognostic factors. Onkologie. 2012;35(4):210-4. http://doi.org/bt92.
8.Smith MB, Silverman JF, Raab SS, Towell BD, Geisinger KR. Fine-needle aspiration cytology of hepatic leiomyosarcoma. Diagn Cytopathol. 1994;11(4):321-7. http://doi.org/dx4gk9.
9.Sprogøe-Jakobsen S, Hølund B. Immunohistochemistry (Ki-67 and p53) as a tool in determining malignancy in smooth muscle neoplasms (exemplified by a myxoid leiomyosarcoma of the uterus). APMIS. 1996;104(10):705-8. http://doi.org/dgvwjf.
10.Reichel C, Fehske W, Fischer HP, Hartlapp JH. Undifferentiated (embryonal) sarcoma of the liver in an adult patient with metastasis of the heart and brain. Clin Investig. 1994;72(3):209-12. http://doi.org/bqtws7.
11.Holloway H, Walsh CB, Thomas R, Fielding J. Primary hepatic leiomyosarcoma. J Clin Gastroenterol.1996;23(2):131-3. http://doi.org/b4277c.
12.Brichard B, Smets F, Sokal E, Clapuyt P, Vermylen C, Cornu G, et al. Unusual evolution of an Epstein-Barr virus-associated leiomyosarcoma occurring after liver transplantation. Pediatr Trasplant. 2001;5(5):365-9. doi:. http://doi.org/cphh9w.
13.Jyh-Wei C, Gin-Ho L, Kwok-Hung L, Hung-Tai C, Huay-Ben P, Hui-Hwa T. Primary malignant fibrous histiocytoma of the liver: report of a case. Gastroenterol-Taiwan. 1995;12:316-21.
14.Forbes A, Portmann B, Johnson P, Williams R. Hepatic sarcomas in adults: a review of 25 cases. Gut. 1987;28(6):668-74. http://doi.org/c55rhc.
15.Zornig C, Kremer B, Henne-Bruns D, Weh HJ, Schröder S, Brölsch CE. Primary sarcoma of the liver in the adult. Report of five surgically treated patients. Hepatogastroenteroly. 1992;39(4):319-21.
16.Moreno A, Vicente M, Del Pozo M, Sola J, Jiménez A. Sarcoma indiferenciado de hígado: a propósito de un caso con diagnóstico preoperatorio. Cir Esp. 1995;58:68-70.
17.Miettinen M, Kahlos T. Undifferentiated (embryonal) sarcoma of the liver. Epithelial features as shown by inmunohistochemical analysis and electron microscopic examination. Cancer. 1989;64(10):2096-103. http://doi.org/fbtcnz.
18.Fujita H, Kiriyama M, Kawamura T, Ii T, Takegawa S, Dohba S, et al. Primary hepatic leiomyosarcoma in a woman after renal transplantation: report of a case. Surg Today. 2002;32(5):446-9. http://doi.org/dst86c.
19.Pinson CW, Lopez RR, Ivancev K, Ireland K, Sawyers JL. Resection of primary hepatic malignanant fibrous histiocytoma, fibrosarcoma and leiomyosarcoma. South Med J. 1994;87(3):384-91. http://doi.org/dnffr8.
20.Botella MT, Cabrera T, Sebastián JJ, Navarro MJ, Álvarez R, Uribarrena R. Hemangioendotelioma epiteloide hepático: un raro tumor hepático. Rev Esp Enferm Dig. 1995;87(10):749-51.
21.Marín R, Cabello A, Bondía JA, Moreno FJ, Ribeiro M, Fernández JL, et al. Seudotumor inflamatorio de hígado: presentación de un nuevo caso. Cir Esp. 1997;62:253-4.
22.Martínez Isla A, Ferrara A, Badia JM, Holloway I, Tanaka H, Riaz A, et al. Hepatocarcinoma fibrolamelar: resultados de la resección hepática parcial. Rev Esp Enferm Dig. 1997;89(9):699-705.
23.Aller R, Moreira V, Bermejo F, Sanromán AL, de Luis DA. Leiomiosarcoma de vena cava inferior: Aproximación diagnóstica y terapéutica. Rev Esp Enf Digest. 1997;89(9):706-10.
24.Das Gupta TK, Patel MK, Chaudhuri PK, Briele HA. The role of chemotherapy as an adjuvant to surgery in the initial treatment of primary soft tissue sarcomas in adults. J Surg Oncol. 1982;19(3):139-44.
25.Alrenga DP. Primary fibrosarcoma of the liver. Case report and review of the literature. Cancer. 1975;36(2):446-9. http://doi.org/b2mng4.
26.Higuchi T, Kikuchi M, Yamada Y. Rapidly growing hepatic leiomyosarcoma: an immunohistochemical evaluation of malignant potential with monoclonal antibody MIB-1. Am J Gastroenterol. 1994;89(11):2098-9.
27.King ME, Dickersin GR, Scully RE. Myxoid leiomyosarcoma of the uterus. A report of six cases. Am J Surg Pathol. 1982;6(7):589–98.
28.Gates LK, Cameron AJ, Nagorney DM, Goellner JR, Farley DR. Primary leiomyosarcoma of the liver mimicking liver abscess. Am J Gastroenterol. 1995;90(4):649-52.
29.Shivathirthan N, Kita J, Iso Y, Hachiya H, Kyunghwa P, Sawada T, et al. Primary hepatic leiomyosarcoma: Case report and literature review. World J Gastrointest Oncol. 2011;3(10):148-52. http://doi.org/dzqsj2.
30.Oosten AW, Seynaeve C, Schmitz PI, den Bakker MA, Verweij J, Sleijfer S. Outcomes of first-line chemotherapy in patients with advanced or metastatic leiomyosarcoma of uterine and non-uterine origin. Sarcoma. 2009;2009: 348910. http://doi.org/cbmrhh.
31.Husted TL, Neff G, Thomas MJ, Gross TG, Woodle ES, Buell JF. Liver transplantation for primary or metastatic sarcoma to the liver. Am J Transplant. 2006;6(2):392-7.