Network of Scientific Journals from Latin America and the Caribbean, Spain and Portugal
Fisicoquímica e Inorgánica
Synthesis, characterization, X-ray crystal structure and DFT calculations of 4-([2,2':6',2''-terpyridin]- 4'-yl)phenol
Síntesis, caracterización, estructura cristalina y cálculos DFT para el 4-([2,2':6',2''-terpiridin]-4'-il)fenol
Síntese, caracterização, estrutura de cristal e cálculos DFT para o 4-([2,2':6',2''-terpiridina]-4'-il)fenol
Synthesis, characterization, X-ray crystal structure and DFT calculations of 4-([2,2':6',2''-terpyridin]- 4'-yl)phenol
Revista Colombiana de Química, vol. 47, no. 1, 2018
Universidad Nacional de Colombia
Received: 17 June 2017
Accepted: 25 September 2017
Abstract: The synthesis of new terpyridine (Tpy) derivatives has been subject of extensive research due to its potential as functional materials for solar energy conversion, among other applications. In this contribution, the 4-([2,2':6',2''-terpyridin]-4'-yl)phenol (TpyOH) has been synthesized, characterized and studied through several methods, including X-ray crystallography and computational approaches. Single crystal X-ray structure analysis shows that TpyOH is essentially planar, with dihedral angles of about 5.03° between the central pyridinyl and the phenolic ring, and also 6.05 and 12.2° in the terpyridine moiety. In the crystal, molecules are linked by intermolecular hydrogen bonds and through π-π stacking interactions. Using a time-dependent density functional theory approach and taking into account bulk solvent effects, the absorption and fluorescence spectra of TpyOH were investigated and compared. The TD-DFT S0 --> Sn and S1 --> S0 transition energies are in good agreement with experimental results. The frontier molecular orbitals analysis showed that the low-energy absorption band has an intraligand charge transfer character (ICT), while the high-energy band is a common feature of p–p* transitions of the Tpy moiety. The S1 --> S0 emission transition also has an ICT character, with a 90% contribution from the HOMO --> LUMO transitions.
Keywords: Terpyridine, Krönhke reaction, Crystal structure, TD-DFT.
Resumen: La síntesis de derivados terpiridinicos (Tpy) se ha investigado ampliamente debido a su potencial para la conversión de energía solar. En este artículo se sintetizó y caracterizó el 4-([2,2':6',2''-terpiridin]-4'-il)fenol (TpyOH), a través de varias metodologías como la cristalografía de rayos X y herramientas computacionales. El análisis de rayos X de monocristal mostró que el TpyOH es plano, con ángulos diedros de 5,03° entre el piridinilo central y el anillo fenólico, con presencia de ángulos de 6,05 y 12,2º en la porción terpiridínica. En el cristal, las moléculas están unidas por enlaces de hidrógeno intermoleculares y mediante interacciones de apilamiento π-π. Utilizando cálculos DFT dependientes del tiempo (TD-DFT) y teniendo en cuenta el efecto de los disolventes, se investigaron y compararon los espectros de absorción y fluorescencia de TpyOH. Las energías de transición TD-DFT de S0 --> Sn y S1 --> S0 concuerdan con los resultados experimentales. El análisis de orbitales moleculares de frontera mostró que la banda de absorción de baja energía corresponde a transferencia de carga intraligando (ICT); mientras que la banda de alta energía es común en las transiciones p–p* del resto Tpy. La emisión debido a la transición S1 --> S0 corresponde a ICT, con una contribución del 90% proveniente de transiciones HOMO --> LUMO.
Palabras clave: Terpiuridina, reacción de Krönhke, estructura cristalina, TD-DFT.
Resumo: A síntese de derivados de terpiridina (Tpy) tem sido estudada devido ao seu potencial para a conversão de energia solar. Nesta contribuição, o 4-([2,2':6',2''- terpiridina]-4'-il) fenol (TpyOH) foi sintetizado, caracterizado e estudado por vários métodos. A análise de estrutura de raios X de cristal único mostra que o TpyOH é plano, com ângulos diedros de 5,03 ° entre o piridinilo central e o anel fenólico, e também 6,05 e 12,2 ° na porção de terpiridina. No cristal, as moléculas são ligadas por ligações intermoleculares de hidrogênio e através de interações de empilhamento π-π. Usando uma abordagem da teoria funcional da densidade dependente do tempo e levando em consideração os efeitos do solvente em massa, foram investigados e comparados os espectros de absorção e fluorescência do TpyOH. As energias de transição TD-DFT S0 --> Sn e S1 --> S0 estão de acordo com os resultados experimentais. A análise de orbitários moleculares de fronteira mostrou que a banda de absorção de baixa energia possui um caráter de transferência de carga intraligando (TIC), enquanto a banda de alta energia é uma característica comum das transições p–p*da fração Tpy. A transição de emissão S1 ---> S0 também tem um caráter TIC, com uma contribuição de 90% das transições HOMO®LUMO.
Palavras-chave: Terpiridine, reação de Krönhke, estrutura de cristal, TD-DFT.
Introduction
Currently, terpyridines (Tpys) have a very important presence in the scientific literature due principally to their optoelectronic properties and coordination capability (1, 2, 3). The different characteristics of Tpys are provided by the three neighbouring pyridine rings and their spatial disposition, which form a very particular and changeable electronic space. As a result, for example, Tpys are capable of forming coordination compounds with various transition metals. This makes Tpys very promissory moieties in the development of new materials with high technological potentials like photovoltaics (4), LEDs (5), solar cells (6, 7, 8, 9) and chemo-sensor probes (10).
This interest in Tpys has represented a considerable effort on the synthetic field. Many methodologies have been widely studied for the synthesis of Tpys, like the Potts, Jameson, Sasaky, Stille cross-coupling, and the Kröhnke reaction (11), the latter being the most useful methodology to synthesize 4-aryl substituted Tpys using very simple reagents under feasible conditions. The Kröhnke reaction involves the use of the appropriate aryl-aldehydes and 2-acetylpyridine to give the corresponding 4-substituted Tpy in moderate yields. This is a very important advantage over the aforementioned methodologies which require very expensive reagents non-commercially available. However, the Kröhnke methodology has a very important limitation regarding the synthesis of Tpys, since this synthetic route can only be used for the synthesis of 4-aryl substituted Tpys.
The spatial disposition of the nitrogen atoms over the pyridine rings of Tpys is very important for the physical and chemical properties. As a result of the free rotation presented by the sigma bonds that connect the aromatic rings, Tpys have different structural conformations, which can present high or low π-electronic delocalization, properties that can affect the optoelectronic properties drastically. Interestingly, the conformation in Tpys can be altered or modified by specific guest, such as metal ions, through coordination bonds. Therefore, correlation of spatial conformations, crystallographic structure, and optoelectronic properties of Tpys are very important for technological applications such as chemical sensors. Herein, the synthesis, characterization, and the crystal structure of 4-([2,2':6',2''-terpyridin]-4'-yl)phenol (TpyOH) are reported, a versatile tridentate ligand which may be used as building-block for the synthesis of multicomponent systems with interesting photochemical and photophysical properties.
Materials and methods
Synthesis
4-Hydroxybenzaldehyde and 2-acetylpyridine were purchased from Sigma-Aldrich (St. Louis - MO, USA) and were used without further purification. Sodium hydroxide was obtained from Alfa-Aesar (Haverhill - MA, USA). Following the Kröhnke reaction (Figure 1), TpyOH was obtained in moderate yield.
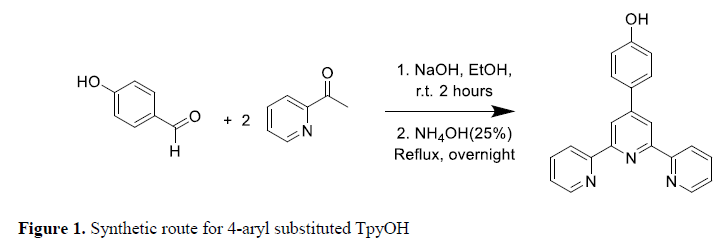
Synthetic route for 4-aryl substituted TpyOH
General procedure
Synthesis of 4-([2,2':6',2''-terpyridin]-4'-yl)phenol (TpyOH)
2.41 mL of 2-acetylpyridine (20 mmol) were added dropwise to a solution of 1.3 g of 4-hydroxybenzaldehyde (10 mmol) and 2.1 g of sodium hydroxide (50 mmol) in 25 mL of ethanol at 0 ºC. Two hours later having reached the room temperature, 10 mL of concentrated ammonium hydroxide were added and the solution was refluxed overnight. After this, filtration at room temperature gave a yellowish solid that was recrystallized from ethanol-water (1:1) to yield 2.03 g (57%) of light yellow crystals. M.p. > 300 °C (lit. (11) 302-306ºC). FT-IR (cm-1): 3441, 3055-3039, 1597, 1527 and 1095. 1H-RMN (400 MHz, CDCl3) δ (ppm): 8,79 (d, 2H), 8,75 (d, 2H), 8,70 (s, 2H), 7,72–7,78 (m, 2H), 7,38–7.41 (m, 6H). 13C-RMN (100 MHz, CDCl3) δ (ppm): 116.52, 117.53, 121.39, 125.02, 128.31, 128.58, 139.12, 149.71, 155.59, 160.13 MW: Calculated: 325.1201 Found: 325.171(MALDI-TOF).
Steady-state spectroscopy
All measurements were performed at room temperature (20 ± 1°C) in HPLC quality ethanol. The stationary absorption spectra were measured using a Shimadzu UV-2401 PC double channel spectrophotometer (Kyoto, Japan) in a 1 cm quartz cell. The steady-state fluorescence emission and excitation spectra were recorded using a PTI Quantamaster™ 4000 spectrofluorometer (New Jersey, USA).
Computational details
All calculations were performed using the Gaussian09 quantum chemical program package (12). The molecular structures of TpyOH in the electronic ground state were fully optimized with density functional theory (DFT) method using the parameter-free PBE0 hybrid functional and the 6-31+G(d,p) basis sets. To include the solvent effects, we used the Polarized Continuum Model (PCM) with the integral equation formalism (IEFPCM) developed by Tomasi et al (13, 14).
To simulate the absorption and emission spectra, vertical excitation energies in the singlet and triplet manifold were calculated using the PCM/TD-PBE0/6-31+G(d,p) level of theory. This approach has proven to predict accurate descriptions of excited states for several organic (15) and metal-organic complexes (16, 17, 18). Geometry optimizations for the S0 and S1 states were performed without symmetry constraints and were confirmed to be stationary points through vibrational frequency analysis.
Results and discussion
Synthesis and crystal structure
TpyOH was synthesized under mild conditions following the Kröhnke reaction with a yield similar to the previously reported (11). After purification, TpyOH was crystallized in an orthorhombic crystal system with P212121 symmetry. Full information of the structure was deposited at the CSD Database under the deposition number 1454127 (Table 1). The crystal-packing diagram (Figure 2) shows that the asymmetric unit of TpyOH contains one crystallographically independent molecule as part of four molecules per unit cell with similar bond distance and dihedral angles. The geometry of TpyOH corresponds to a nearly coplanar structure, where the central pyridinyl ring (Py2) makes a dihedral angle of about 5.03° with the phenolic ring (PhOH), and 6.05 and 12.2° with the pyridinyl substituent rings Py1 and Py3, respectively.
The differences in the dihedral angles observed on the terpyridyl moiety can be attributed to the intermolecular hydrogen bond O25-H26××××N18 formed between two adjacent molecules. As shown in Figure 1, the molecules are column-like stapled along the a-axe of the unit cell with a packing dominated mainly by intermolecular hydrogen bonds (1.85(3) Å), between the pyridinyl nitrogen of Py3 and the –OH over the phenol-group at the neighbour molecule and by π-p interactions (3.52(3) Å) between molecules of the same staple. Although the crystal structure of the target compound was published earlier (19) and the crystallographic data are quite similar, the cell volume here reported is a little bit smaller (about 68 Å), showing the possibility of polymorph formation for this compound (20).
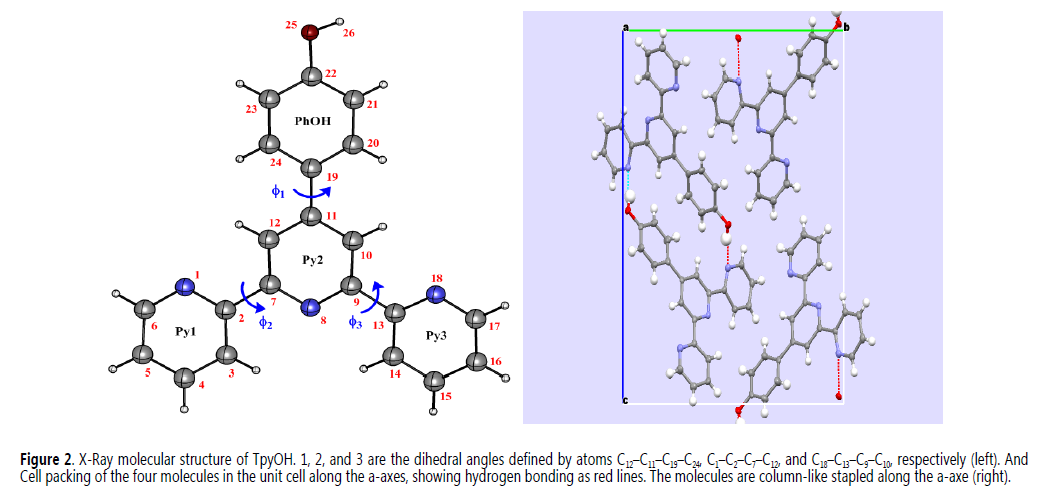
X-Ray molecular structure of TpyOH. f1, f2, and f3 are the dihedral angles defined by atoms C12–C11–C19–C24, C1–C2–C7–C12, and C18–C13–C9–C10, respectively (left). And Cell packing of the four molecules in the unit cell along the a-axes, showing hydrogen bonding as red lines. The molecules are column-like stapled along the a-axe (right)

Crystal data and structure refinement for TpyOH
Molecular Hirschfeld surface
The Hirshfeld surface is a useful tool for describing the surface characteristics of molecules. The molecular Hirshfeld surface of TpyOH was generated by the CrystalExplorer 17.5 program (21), using a standard high surface resolution with the 3D dnorm surface (Figure 3) mapped over a fixed color scale of -0.66 (red) to 1.2 Å (blue). The Hirshfeld surface is shown to be transparent to permit visualization of the molecular moiety, and the 3D dnorm surface was used to identify close intermolecular interactions. The values of dnorm are negative or positive either the intermolecular contacts are shorter or respectively longer than the Van der Waals radii. The dnorm values were pictured onto the Hirshfeld surface using a red-blue-white color scheme, where red regions represent a closer contact with a negative dnorm value, a blue region represents longer contacts with a positive dnorm value, and a white region represents exactly the distances by van der Waals radii with a dnorm value of zero.
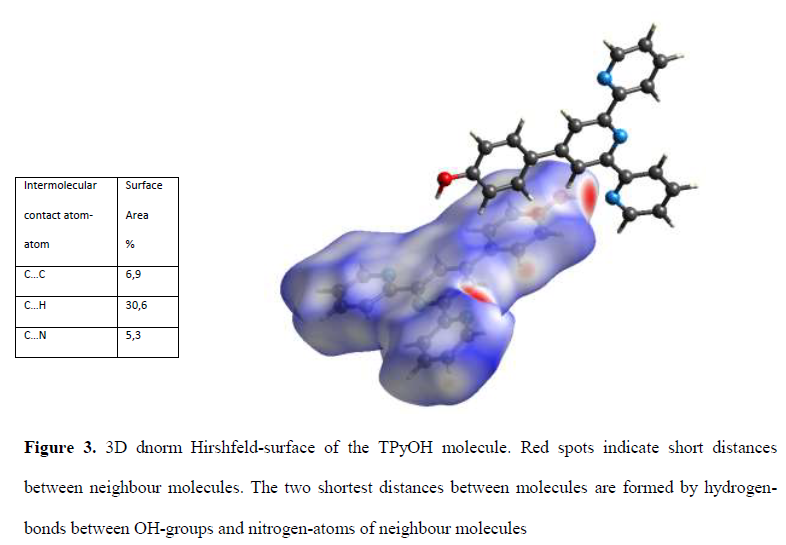
3D dnorm Hirshfeld-surface of the TPyOH molecule. Red spots indicate short distances between neighbour molecules. The two shortest distances between molecules are formed by hydrogen-bonds between OH-groups and nitrogen-atoms of neighbour molecules
The 2D fingerprint plots (Figure 4) can be interpreted as particular atom pair contacts between neighbour molecules. It enables the separation of different interaction types that overlap in the full fingerprint (Figure 4a). The 2D fingerprints are symmetrical along the diagonal. The de- and di-axes represent the distances from the molecule surface to the nearest nucleus external to the surface (de) and the distance from the surface to the nearest nucleus internal to the surface (di). These values provide an immediate picture of the nature of intermolecular contacts in the crystal.
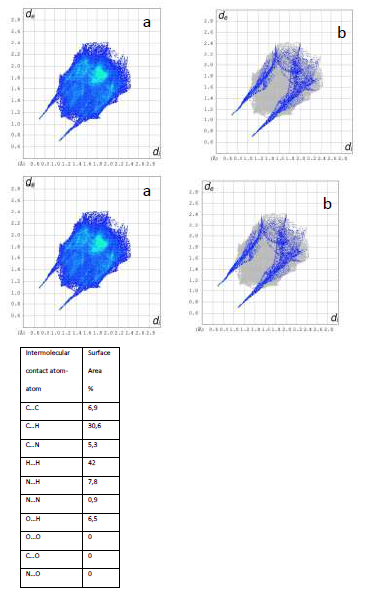
In Figure 4b the spikes represent the nearest contact between N- and H-atoms (di=1.008Å, de=0.730 Å) but in this type of interaction only 7.8% of the Hirshfeld surface is involved, the main contact area between the surrounding molecules is formed between H--H (42% of the surface) and between C--H atoms (30.6% of the surface).
Steady-state spectroscopy
The absorption and emission spectra of TpyOH in ethanol solution are shown in Figure 5. As can be observed, the absorption spectrum shows an intense band centred at λ = 286 nm and a weak long-wavelength absorption in the 330–410 nm range. The low-energy band is assigned to the intraligand charge transfer transition (ICT), while the high-energy band is a common feature of p–p* transitions of the Tpy moiety (22, 23, 24, 25). From the comparison between the UV-visible spectrum of TpyOH and the corresponding terpyridine systems bearing electron-donating or electron-withdrawing groups, is clear that the intensity of the ICT band is mainly governed by the electron donating strength of the substituent. Specifically, terpyridines carrying an -NR2 electron donor substituent show a strong absorption band around 360-399 nm.
On the contrary, for the case of NO2 substitution, the spectrum shows only one absorption band at higher energy, as expected for a p–p* transition centred on the ligand (22, 23, 24, 25). Furthermore, the emission spectrum of TpyOH shows a broad emission band centred at λ = 384 nm, with a very large Stokes shift of almost 100 nm, indicating that TpyOH undergoes with difficulty self-absorption processes. This suggests that TpyOH could be a good candidate for applications such as chemo-sensors. This emission band corresponds to energy relaxation from a nearly coplanar S1 state formed after the photo-absorption process (as it is shown in the following Figure 5).
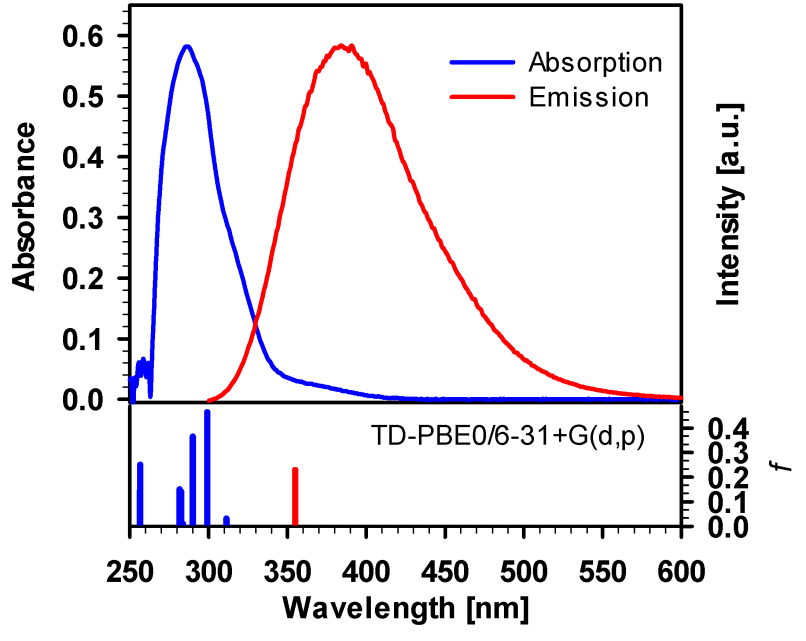
Absorption (blue line) and emission (red line) spectra of TpyOH in ethanol. The fluorescence spectrum was taken with an excitation wavelength of 285 nm. The vertical sticks correspond to the vertical absorption and emission transitions calculated at the PCM/TD-PBE0/6-31+G(d,p) level of theory
DFT ground state properties and conformational analysis
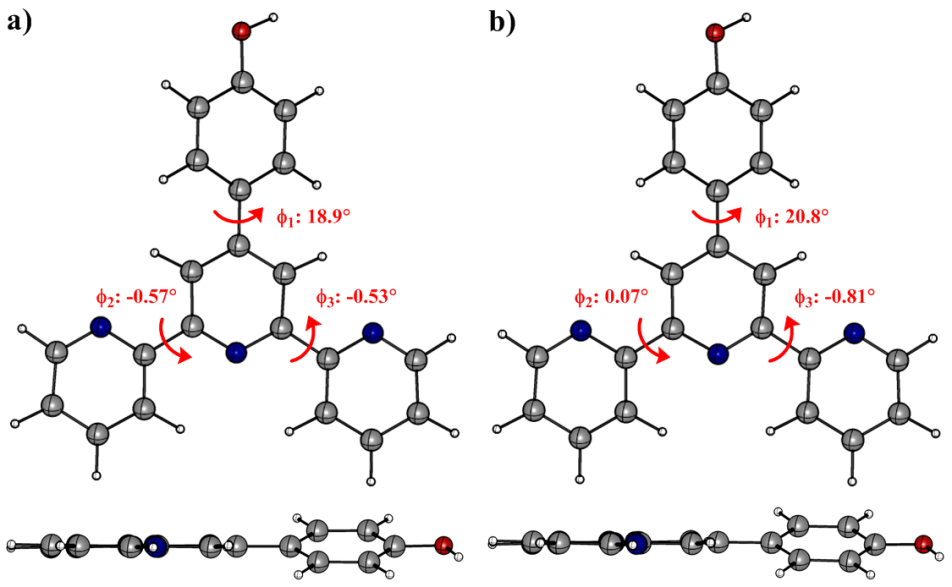
DFT geometry optimization of TpyOH in the ground state was performed taking into account three different isomers, generally named as transoid, cisoid, and a mixture between them (from now on labelled as TpyOH 1, TpyOH 2, and TpyOH 3), which differ in the spatial orientation of the terpyridyl nitrogen atoms. Optimized ground state structures of these Tpy isomers in ethanol are depicted in Figure 6, while selected structural parameters are included in Table 2. For the TpyOH 1 isomer, the phenol substituent forms a dihedral angle of approximately 34° relative to central terpyridine ring, while the three terpyridine aromatic rings are nearly planar, with f2 and f3 of about -1.8° and -1.9°, respectively. For the TpyOH 2 isomer, the substituted phenol ring and the terminal terpyridine rings are twisted for about 38° and -139°, respectively, while for the TpyOH 3 corresponding dihedral angles (see Figure 2 for angles designations) are f1 = 36°, f2 = -144° and f3 = -3.7°.
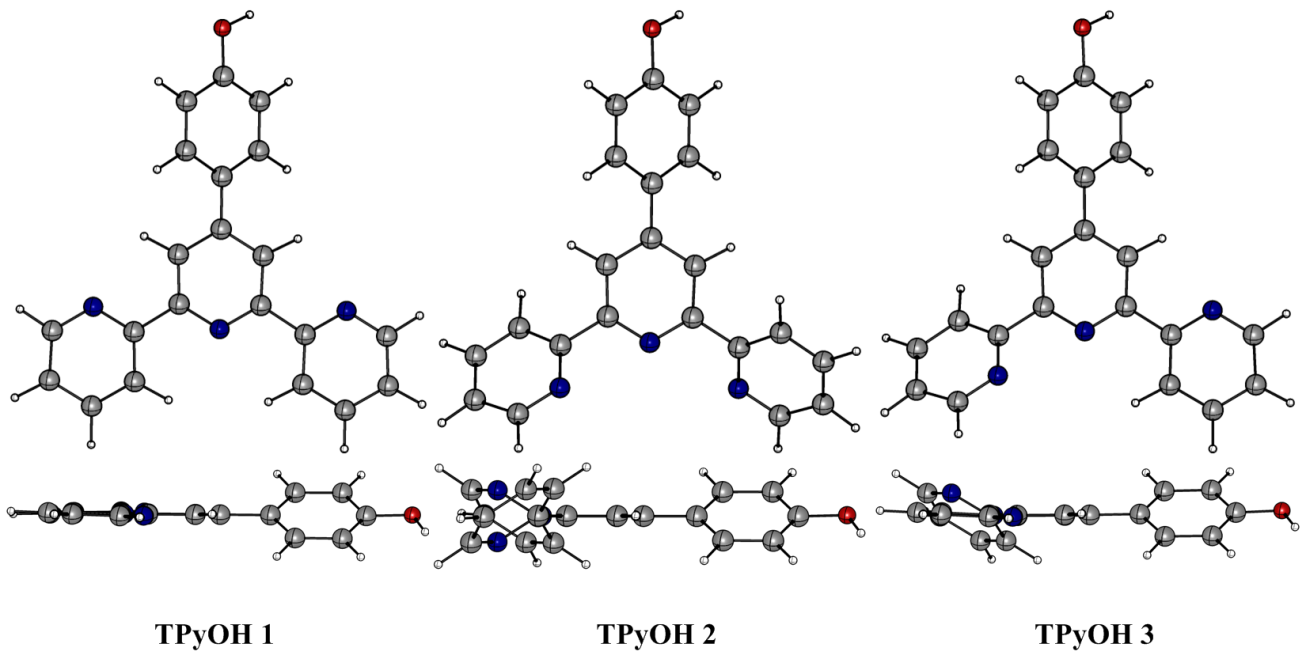
Optimized molecular structures of TpyOH 1, TpyOH 2 and TpyOH 3 conformers in ethanol, calculated at the PCM/TD-PBE0/6-31+G(d,p) level of theory
The results showed that TpyOH 1 is the most stable conformer, with the TpyOH 2 and TpyOH 3 conformers lying higher in energy at 13.0 and 5.51 kcal/mol in the gas phase, and 6.52 and 3.12 kcal/mol in ethanol. Comparing Figures 2 and 6, it is clear that TpyOH 1 corresponds to the crystal structure, for this reason, in the following section, we will only limit the analysis to this conformer.
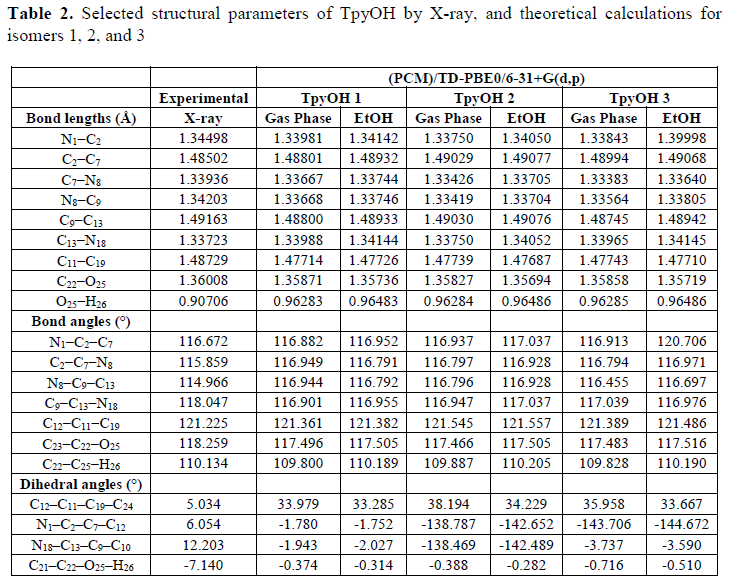
Selected structural parameters of TpyOH by X-ray, and theoretical calculations for isomers 1, 2, and 3
S0 --> Sn excitation energies calculations. Absorption spectrum
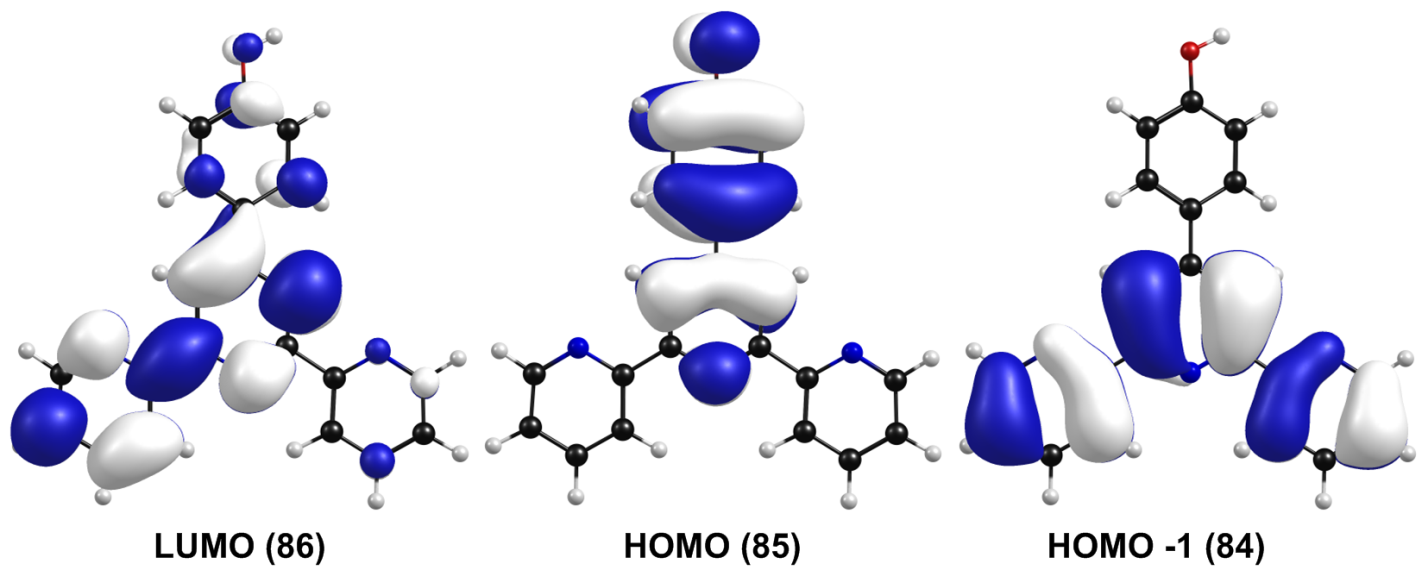
TD-DFT computational studies were performed to elucidate the electronic structures of the S0 state of TpyOH 1. The calculated excitation energies with their contribution, oscillator strengths, and coefficients are reviewed in Table 3. The best agreement with the experimental data was obtained using the PCM/TD-PBE0/6-31+G(d,p) level of theory. Specifically, the calculated S0 --> S1 ICT transition is 3.98 eV (311 nm), while the S0 --> S2 transition is 4.15 eV (299 nm). This table also shows that the lowest-energy singlet --> singlet transition at 311 nm has its main contribution from the highest occupied molecular orbital HOMO (DFT orbital 85) to the lowest unoccupied molecular orbital LUMO (DFT orbital 86) and is assigned as an ICT transition.
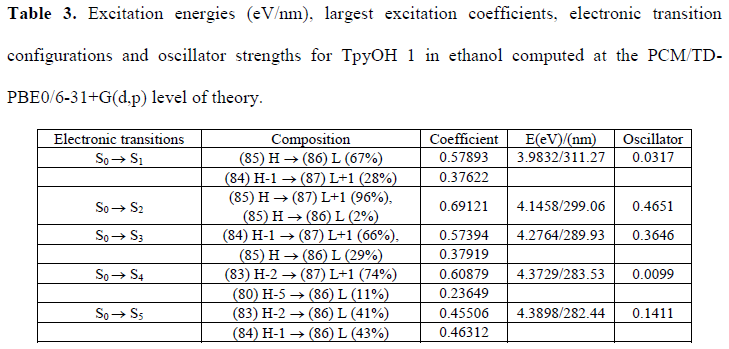
Excitation energies (eV/nm), largest excitation coefficients, electronic transition configurations and oscillator strengths for TpyOH 1 in ethanol computed at the PCM/TD-PBE0/6-31+G(d,p) level of theory.
The frontier molecular orbitals and its composition are shown in Figure 7 and Table 4, respectively. From Table 4, on the one hand, it can be observed that the HOMO is composed of 34.0% p (Tpy) and 66.0% p (PhOH), whereas the LUMO orbital is localized on the terpyridine moiety with a 99.0% π* (Tpy). On the other hand, the next high-energy transitions at 299 nm (f = 0.4651, Table 3) and 289 nm (f = 0.3646, Table 3) dominate the energy absorption band. Table 3 also shows that the S2 state has its main contribution from the H --> L+1 transitions (96%), while the S3 state has contributions from the H --> L ICT transitions (29%) mixed with the H-1 --> L+1 (66%) transition. Both states have a p–p* character due to the p–p* transitions of the terpyridine and the phenol ring.
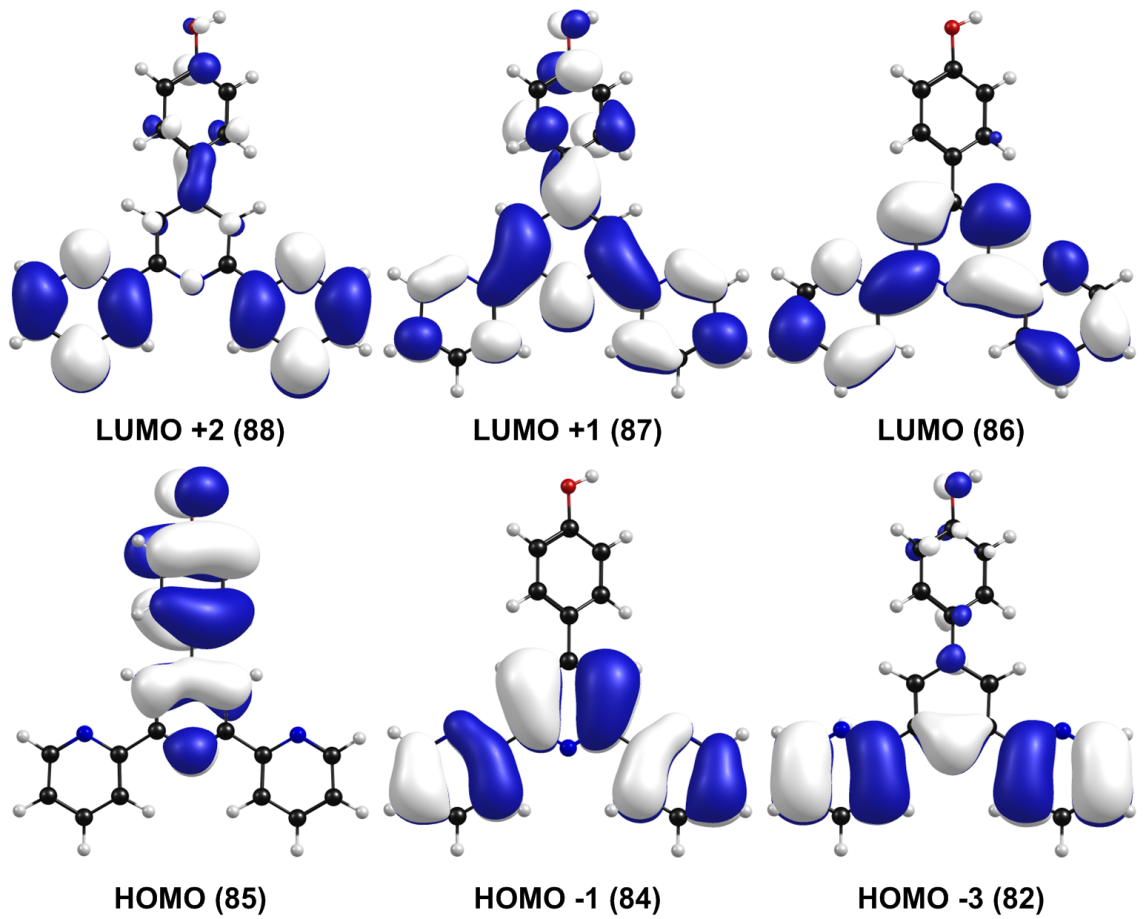
Frontier molecular orbitals of TpyOH 1 in ethanol calculated at the PCM/TD-PBE0/6-31+G(d,p) level of theory (isosurface value: 0.02)
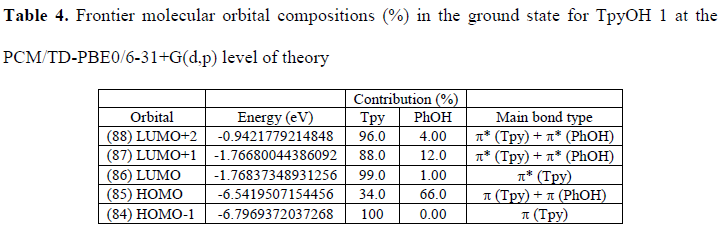
Frontier molecular orbital compositions (%) in the ground state for TpyOH 1 at the PCM/TD-PBE0/6-31+G(d,p) level of theory
Optimization of the S1 excited state. Fluorescence emission spectrum
Optimization of the S1 Franck−Condon state of TpyOH 1 in both gas phase and ethanol solution resulted in a more coplanar structure (Figure 8). Specifically, the dihedral angles f1, f2 and f3 decrease 15.1, 1.19, and 1.41° in the gas phase, and 12.5, 1.68, and 1.21° in ethanol, respectively. According to these results, we can conclude that the resonance interaction is more efficient in the first excited state than in the ground state. Table 5 lists the calculated emission energy, electronic transition configuration, and frontier molecular orbital compositions (%) in the S1 excited state of TpyOH 1. As can be observed, the calculated S1 --> S0 transition at 355 nm in ethanol has its main contribution from the HOMO and LUMO orbitals (90% contribution) and oscillator strength of 0.2383. This result shows the ICT character of the emission band.
In the S1 excited state, the HOMO is composed of 35.0% p (Tpy) and 65.0% p (PhOH), whereas the LUMO orbital is mainly localized on the terpyridine moiety with a 94.0% π* (Tpy). The frontier molecular orbitals in the S1 excited state of TPyOH 1 in ethanol are depicted in Figure 9.
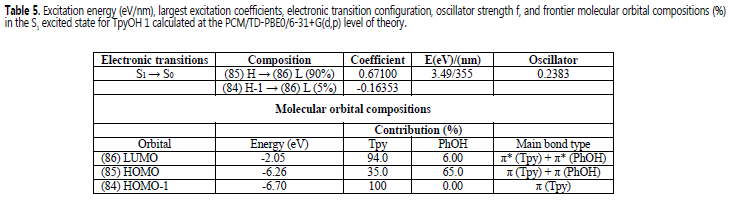
Excitation energy (eV/nm), largest excitation coefficients, electronic transition configuration, oscillator strength f, and frontier molecular orbital compositions (%) in the S1 excited state for TpyOH 1 calculated at the PCM/TD-PBE0/6-31+G(d,p) level of theory
Frontier molecular orbitals in the S1 excited state of TPyOH 1 in ethanol calculated at the PCM/TD-PBE0/6-31+G(d,p) level of theory (isosurface value: 0.02).
Conclusions
In this contribution, the 4-([2,2':6',2''-terpyridin]-4'-yl)phenol compound was synthesized in a moderate yield following the Krönhke conditions and its structure was unambiguously determined by X-ray single-crystal diffraction, 1H- and 13C-NMR, and UV-vis techniques. Crystallographic and DFT results showed that the transoid conformation (TpyOH 1 isomer) corresponds to the most stable and predominant structure for the model compound. Comparison between the geometrical parameters of the optimized ground state and the crystal data shows that the differences are related to the intermolecular hydrogen bond formed between two adjacent molecules in the crystal packing.
The S0 --> Sn and S1 --> S0 transition energies calculated with the PCM/TD-PBE0/631+G(d,p) level of theory are in good agreement with experimental results. TD-PBE0 results show that both the absorption band observed in the 330–410 nm range and the emission band have their main contribution from the HOMO to the LUMO orbitals and are assigned as intraligand charge transfer (ICT) transitions.
Acknowledgments
For financial support, we are thankful to Colciencias (Grants 534-2011 and 111865843739) and to Direccion Nacional de Investigaciones-UNAL (Grant 18296).
References
1. Sun, Y.; El Ojaimi, M.; Hammitt, R.; Thummel, R. P.; Turro, C. Effect of Ligands with Extended π-System on the Photophysical Properties of Ru(II) Complexes. Phys. Chem. B. 2010, 114, 14664-14670. DOI: https://doi.org/10.1021/jp102613n.
2. Schubert, U. S.; Winter, A.; Newkome, G. R. Terpyridine-based Materials: For Catalytic, Optoelectronic and Life Science Applications. Wiley-VCH. Weinheim, Alemania, 2011; pp 260-280. DOI: https://doi.org/10.1002/9783527639625
3. Schubert, U. S.; Hofmeier, H.; Newkome, G. R. Modern Terpyridine Chemistry. Wiley-VCH. Weinheim, Alemania, 2006; pp 26-41. DOI: https://doi.org/10.1002/3527608486.
4. Duprez, V.; Biancardo, M.; Spanggaard, H.; Synthesis of Conjugated Polymers Containing Terpyridine−Ruthenium Complexes: Photovoltaic Applications. Macromolecules. 2005, 38, 10436-10448. DOI: https://doi.org/10.1021/ma051274f.
5. Holder, E.; Marin, V.; Meier, M.; Schubert, U.; A Novel Light-Emitting Mixed-Ligand Iridium(III) Complex with a Terpyridine-Poly(ethylene glycol) Macroligand. Macromolecular Rapid Comm. 2004, 25, 1491-1496. DOI: https://doi.org/10.1002/marc.200400206.
6. Knoll, J. D.; Albani, B. A.; Turro, C.; New Ru(II) Complexes for Dual Photoreactivity: Ligand Exchange and 1O2 Generation. Acc. Chem. Res. 2015, 48, 2280-2287. DOI: https://doi.org/10.1021/acs.accounts.5b00227.
7. Yang, S. H.; Wu, K. L.; Chi, Y.; Cheng, Y. M.; Chou, P. T.; Tris(thiocyanate) ruthenium(II) sensitizers with functionalized dicarboxyterpyridine for dye-sensitized solar cells. Angew Chem. Int. Ed. Engl. 2011, 50, 8270-8274. DOI: https://doi.org/10.1002/anie.201103515.
8. Li, F.; Jiang, Y.; Zhang, B.; Huang, F.; Gao, Y.; Sun, L.; Towards A Solar Fuel Device: Light-Driven Water Oxidation Catalyzed by a Supramolecular Assembly. Angew Chem. Int. Ed. 2012, 51, 2417-2420. DOI: https://doi.org/10.1002/anie.201108051.
9. Lin, R. G.; A new zinc(II) coordination compound of meta-aminobenzoate and 4,4′-bipyridine with broad-band photoluminescence emission. Inorg. Chim. Acta. 2015, 432, 46-49. DOI: https://doi.org/10.1016/j.ica.2015.03.03.
10. Thornleya, P.; Starkeya, J.; Zibasereshta, R.; Polsona, M.; Wikairaa, M. and Hartshorna, R. 4′-(o-Toluyl)-2,2′ : 6′,2′′-terpyridine: synthesis, bromination, complexation, and X-ray crystallographic characterization. J. Coord. Chem. 2011, 64, 145-158. DOI: https://doi.org/10.1080/00958972.2010.546397.
11. Anthonysamy, A.; Balasubramanian, S.; Shanmugaiah, V.; Mathivanan, N. Synthesis, characterization and electrochemistry of 4′-functionalized 2,2′:6′,2″-terpyridine ruthenium(II) complexes and their biological activity. Dalton Transactions, 2008, 16, 2136–2143. DOI: https://doi.org/10.1039/b716011a.
12. Frisch, M.; Trucks, G.; Schlegel, GB.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, et al. Gaussian 09 package: Gaussian 09, Revision E.01.Gaussian, Inc., Wallingford CT. 2009.
13. Cances, E.; Mennucci, B.; Tomasi, J.; A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. DOI: https://doi.org/10.1063/1.474659.
14. Mennucci, B.; Cances, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B. 1997, 101, 10506–10517. DOI: https://doi.org/10.1021/jp971959k.
15. Plaza-Medina, E.; Rodríguez-Córdoba, W.; Morales-Cueto, R.; Peon, J. Primary Photochemistry of Nitrated Aromatic Compounds: Excited-State Dynamics and NO· Dissociation from 9-Nitroanthracene. J. Phys. Chem. A. 2011, 115, 577-585. DOI: https://doi.org/10.1021/jp109041y.
16. Velmurugana, G.; Venuvanalingam, P. Luminescent Re(I) terpyridine complexes for OLEDs: what does the DFT/TD-DFT probe reveal? Dalton Trans. 2015, 44, 8529-8542. DOI: https://doi.org/10.1039/c4dt02917h.
17. Zhang, T.; Jia, J.; Ren, Y.; Wu, H. Ligand Effects on Structures and Spectroscopic Properties of Pyridine-2-aldoxime Complexes of Re(CO)3+: DFT/TDDFT Theoretical Studies. J. Phys. Chem. A. 2011, 115, 3174-3181. DOI: https://doi.org/10.1021/jp200872b.
18. Martinez Saavedra, H.; Ragone, F.; Ruiz, G.; Gara, P.; Wolcan, E. Solvent Dependent Switching of 3MLLCT and 1IL Luminescent States in [ClRe(CO)3(bathocuproinedisulfonate)]2 – Spectroscopic and Computational Study. J. Phys. Chem. A. 2014, 118, 9661-9674. DOI: https://doi.org/10.1021/jp506890r.
19. Darabi, F.; Hadadzadeh, H.; Simpson, J.; Shahpiric, A. A water-soluble Pd(II) complex with a terpyridine ligand: experimental and molecular modeling studies of the interaction with DNA and BSA; and in vitro cytotoxicity investigations against five human cancer cell lines. New J. Chem. 2016, 40, 9081-9097. DOI: https://doi.org/10.1039/C6NJ01880G.
20. Gavezzotti, A.; Fillippini, G. Polymorphic forms of organic crystals at room conditions: Thermodynamic and structural implications. J. Am. Chem. Soc. 1995, 117, 12299-12305. DOI: https://doi.org/10.1021/ja00154a032.
21. M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17. University of Western Australia. http://hirshfeldsurface.net. 2017.
22. Tessore, F.; Roberto, D.; Ugo, R.; Pizzotti, M. Terpyridine Zn(II), Ru(III), and Ir(III) Complexes: The Relevant Role of the Nature of the Metal Ion and of the Ancillary Ligands on the Second-Order Nonlinear Response of Terpyridines Carrying Electron Donor or Electron Acceptor Groups. Inorg. Chem. 2005, 44, 8967-8978. DOI: https://doi.org/10.1021/ic050975q.
23. Kanis, D.; Lacroix, P.; Ratner, M.; Marks, T. Electronic Structure and Quadratic Hyperpolarizabilities in Organotransition-Metal Chromophores Having Weakly Coupled.pi.-Networks. Unusual Mechanisms for Second-Order Response. J. Am. Chem. Soc. 1994, 116, 10089-10102. DOI: https://doi.org/10.1021/ja00101a030.
24. Roberto, D.; Ugo, R.; Bruni, S.; Cariati, E.; Cariati, F.; Fantucci, P.; Invernizzi, I. Quadratic Hyperpolarizability Enhancement of para-Substituted Pyridines upon Coordination to Organometallic Moieties: The Ambivalent Donor or Acceptor Role of the Metal. Organometallics 2000, 19, 1775-1788. DOI: https://doi.org/10.1021/om990865p.
25. Bruni, S.; Cariati, E.; Cariati, F.; Porta, F. A.; Quici, S.; Roberto, D. Determination of the quadratic hyperpolarizability of trans-4-[4-(dimethylamino)styryl]pyridine and 5-dimethylamino-1,10-phenanthroline from solvatochromism of absorption and fluorescence spectra: a comparison with the electric-field-induced second-harmonic generation technique. Spectrochim. Acta Part A. 2001, 57, 1417-1426. DOI: https://doi.org/10.1016/S1386-1425(00)00483-2.
Alternative link
https://revistas.unal.edu.co/index.php/rcolquim/article/view/66281 (html)