
ACTUALIZACIÓN
FACTOR NUCLEAR κB (NF-κB): SIGNALOSOMA Y SU IMPORTANCIA EN ENFERMEDADES INFLAMATORIAS Y CÁNCER
Nuclear factor kB (NF-KB): signalosoma and its importance in cancer and inflammatories diseases
Nancy P. Echeverri R1, Ismena Mockus S.2,
1. Estudiante Maestría en Ciencias-Bioquímica, Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, Bogotá
Correspondencia: npecheverrir@unal.edu.co
2. Docente, Departamento de Ciencias Fisiológicas, Facultad de Medicina, Universidad Nacional de Colombia, Bogotá.
Resumen
El factor nuclear κB (NF-κB) es un dímero constituido por proteínas de la familia Rel. El NF-κB se encuentra en el citoplasma unido a proteínas inhibidoras (IkB). Las IkB son fosforiladas por diferentes cinasas que hacen parte del signalosoma como las cinasas de IKKα e IKKP y el modulador esencial de NF-κB (NEMO), la proteína cinasa activadora de mitosis (MAPK o p38) y la cinasa inductora de NF-κB (NIK). Estas cinasas al ser activadas por señales dependientes de citocinas y luz ultravioleta, fosforilan las IkB provocando su ubiquitinación, su degradación por proteosoma y la subsecuente liberación y translocación al núcleo de NF-κB. Recientemente se le ha dado una gran importancia al NF-kB en la vía de señalización desencadenada por estrés oxidativo, estrés genotóxico y daño en el DNA. A diferencia de la vía denominada clásica, en esta ruta ocurre una SUMOilación de NEMO y translocación al núcleo. En el núcleo NEMO interactúa con la proteína de la ataxia telangiectasia mutada (ATM) activada en respuesta a modificaciones en la cromatina y daño en el DNA. El complejo ATM/NEMO es translocado al citoplasma donde la ATM fosforila a las IKK llevando a la ubiquitinación y posterior liberación de NF-κB que es translocado al núcleo. NF-κB desencadena procesos de supervivencia incluyendo el aumento de la transcripción de enzimas antioxidantes como la superóxido dismutasa, catalasa y glutatión. Estas enzimas participan en el control de los niveles de especies reactivas de oxígeno en la célula. La sobreactivación de NF-κB se relaciona con inflamación y cáncer. En la actualidad se desarrolla una búsqueda de fármacos que actúen sobre moléculas del signalosoma de NF-κB, no sólo para el manejo de enfermedades inflamatorias sino también para el uso durante el tratamiento de tumores resistentes a radio y quimioterapia.
Palabras clave: ataxia telangiectasia, cáncer (neoplasia), inflamación, enzimas, antioxidantes.
Summary
The nuclear factor κB (NF-κB) is a dimer conformed by Reí family. NF-κB is found in cytoplasm bound to inhibitor proteins (IkB). IkB are phosphorilated by different kinases who are part of signalosome as IéB kinases (IKKα, IKKP and NF-κB essential modulator or NEMO), the mitogenic activated protein kinase (MAPK or p38) and NF-éB inducer kinase (NIK). These kinases are activated by different cytokines and ultraviolet light, IkB phosphorilated induce their ubiquitination and proteosome degradation subsequently NF-κB reléase and nucleus translocation.
Nowadays, the NF-κB activation by oxidative stress, genotoxic stress and DNA damage pathways. In contrast with the classical pathway, in this pathway there are a SUMOilation and nuclear translocation of NEMO. In nucleus NEMO interact with ataxia telangiectasia muted which is activated by chromatin changes and DNA damage. The complex ATM/NEMO is later translocated to cytoplasm where IKKβ is phosphorilated by ATM bringing to ubiquitination and thus NF-κB releasing which is translocated to nucleus. NF-κB induces survival rising antioxidants enzymes as superoxide dismutase, catalase and glutathione. These enzymes act in the control of oxidative species levels in the cell. NF-κB over expression is related with inflammation and cancer. Nowadays, is development a pharmacological search which can act inhibiting NF-κB signalosome molecules, not only to inflammatory disease whereas to radiotherapy and chemotherapy cancer resistance.
Key words: ataxia telangiectasia, neoplasms, inflammation, enzymes, antioxidants.
Introducción
El factor nuclear kappa B (NF-κB) se descubrió hace aproximadamente 20 años, como una proteína que se une al potenciador de la cadena ligera κ de inmunoglobulinas en las células B (1). Pertenece a la familia de los factores de transcripción NF-κB la cual es ubicua y participa en la respuesta inmune e inflamatoria; en el desarrollo, formación, progresión y apoptosis de tumores (2).
El NF-κB se activa por varios estímulos como lipopolisacáridos bacterianos, ésteres de forbol, virus, estrés oxidativo, luz ultravioleta, radiación ionizante y drogas genotóxicas. Su activación se produce por las vías de señalización del receptor 1 del factor de necrosis tumoral (TNFR1), el receptor 1 de interleuquina 1 (IL-1R1), el receptor similar a Toll (TLR), el receptor de células B (BCR), el receptor de células T (TCR), receptor de linfotoxina β (LTβR), el factor activador de células B (BAFFR) y el clúster de diferenciación 40 (CD40) (3).
El NF-κB se encuentra en el citoplasma en forma inactiva unido a proteínas inhibidoras, las cuales después de un estímulo adecuado son fosforiladas, ubiquitinizadas y degradadas por el proteosoma, permitiendo así la liberación de NF-κB que se transloca al núcleo donde regula la transcripción de genes diana (4). Dependiendo del estímulo, diferentes proteínas señalizan desde la membrana citoplasmática hacia el núcleo. Además se ha observado que noxas como el estrés genotóxico, el daño en el DNA y estrés oxidativo desencadenan vías de señalización que van desde el núcleo hacia el citoplasma y que comprometen al complejo de activación de NF-κB.
Familias de las proteínas NF-κB, IκB E IKKs
Familia NF-κB
El NF-κB forma parte de la familia Rel, constituida por cinco proteínas que contienen dominios homólogos y que forman entre ellas homo o heterodímeros. Estas proteínas son: p50, p52, p65 (RelA), c-Rel y RelB; de éstas sólo las tres últimas tienen el dominio transactivador, indispensable para reconocer el promotor de los genes diana (Tabla 1) (4).
La formación del homodímero o heterodímero es determinante en la actividad transcripcional en los promotores de los genes diana. El heterodímero p50/p65 es el más común y su actividad transcripcional ocurre gracias a la proteína p65 (2). Aunque los homodímeros p50 y p52 no poseen actividad transcripcional, pueden estimular la transcripción cuando se unen a la proteína nuclear parecida al inhibidor de κB (BCL-3) (5).
Las proteínas p50 y p52 se sintetizan como precursores p105 y p100 respectivamente, procesados por el proteosoma, donde se remueve su extremo carboxilo-terminal (C-ter), dando lugar a las formas activas p50 y p52 (6) (Figura 1).
Proteínas IκBs
Los inhibidores de kB (IkBs) constituyen una familia estructuralmente relacionada, conformada por IkBa, IkBb e IkBε, que contienen múltiples repeticiones de ankirina las cuales interactúan con los dominios de localización nuclear de las proteínas de la familia Rel (Rel Homolog Domain; RHD) previniendo su translocación (4)
En los IκBs se observa una región central que contiene repeticiones de ankirina y un dominio amino-terminal (N-ter) regulador que controla la degradación. Los precursores de las proteínas p50 y p52 también se comportan como IéBs gracias a sus repeticiones de ankirina (7).
En células no estimuladas, NF-κB se encuentra principalmente en el citoplasma donde es secuestrado mediante la interacción con estas proteínas inhibidoras (3). Los IκBs, en respuesta a estímulos con agonistas, son degradados por el proteosoma 26S, liberando los dímeros de NF-κB, los cuales se translocan al núcleo y modulan la expresión génica; la actividad transcripcional de ciertos dímeros de NF-κB es además regulada por fosforilación, siendo éste un mecanismo adicional de control para su función (8) (Figura 1).

Proteínas IKKs
La familia de las cinasas de los inhibidores de NF-κB (IKKs) está constituida por IKKα, IKKβ e IKKy o modulador esencial de NF-κB (NEMO). Los inductores de NF-κB, a través de diferentes receptores y proteínas adaptadoras, desencadenan señales que convergen en la activación del complejo IKK (9). Los homodímeros o heterodímeros de IKKα o IKKβ fosforilan a las proteínas IkBcc, IkB(3 e IKBáinduciendo su ubiquitinación y degradación por el proteosoma permitiendo así la liberación de NF-κB que se transloca a núcleo (10).
Aunque otras cinasas están involucradas en la fosforilación de las IκBs, las IKKs se caracterizan por la rapidez de su activación, la acción simultánea sobre ambos residuos de Ser en las IκBs y una preferencia por las Ser en relación a las treoninas (Thr) (11).
IKKα e IKKβ comparten una homología del 50 por ciento de identidad de secuencia, poseen un dominio cinasa en el N-ter, la cremallera rica en leucina (LZ) y un motivo hélice-curva-hélice (HTH). Estas dos cinasas constituyen la subunidad catalítica del complejo IKKα, IKKβ y NEMO (12).
Una tercera IKK es la IKKy o NEMO, un polipéptido de 48 KDa, rico en ácido glútamico (Glu) y glutamina y que forma multímeros mediante los dominios súper enrollado (CC1), CC2, dominio rico en leucina (LZ) y el dedo de Zn (ZF). El CC1 es necesario para la interacción con las cinasas IKKα e IKKβ (13), el CC2 media la oligomerización y el LZ es crítico para la exportación nuclear (14).
NEMO regula la fosforilación de IKKβ con la ayuda de una proteína rica en ácido glutámico, leucina, lisina y serina (ELKS, según la nomenclatura de aminoácidos con una sola letra), permitiendo su activación. A su vez, el inhibidor IKBa es fosforilado en la Ser 32 y Ser 36 por IKKβ activo. Este cambio postraduccional de IKBa sirve de señal para la ubiquitinación en las Lys 19 y 21, llevando a la subsecuente degradación vía proteosoma 26S, quedando así el NF-κB libre y translocándose al núcleo donde puede activar la transcripción de genes de citocinas, quimocinas y moléculas inhibidoras de apoptosis (15) (Figura 1).
Activación de NF-κB
Ruta clásica o canónica
La ruta clásica de activación de NF-κB es inducida por una variedad de mediadores de respuesta inmune innata y adaptativa, tales como citocinas proinflamatorias (TNFa, IL-1p) (16), la activación del receptor similar a Toll (TLR) (17) y los receptores de antígenos (TCR y BCR) (18). Todas estas cascadas de señalización convergen en la activación de las proteínas IKK y en la degradación de IKBa permitiendo la liberación de los heterodímeros p50/ RelA y p50/c-Rel (3).
En la vía de activación clásica el evento característico es la fosforilación de IKBa por el complejo IKKα/p (19). En esta ruta también interviene NEMO, el cual es requerido para la interacción del complejo IKK con las proteínas corriente arriba de la señal (12). En respuesta a la activación por TNFa e IL-1, IKKβ fosforila a IKBa. La activación de IKKβ por estos estímulos requiere de la proteína activadora de mitosis cinasa-cinasa-cinasa (MAP-3K), MAPK/ERK-cinasa reguladora de señales extracelulares (MEKK3) y cinasa activadora del factor de crecimiento transformante p (TAK1) las cuales probablemente fosforilan directamente a IKKβ (20).
Todos estos inductores de NF-κB, a través de diferentes receptores y proteínas adaptadoras, desencadenan señales que convergen en la activación del complejo cinasa IkB (IKK), complejo que incluye a la molécula de andamiaje NEMO y a las IKKα e IKKβ (10).
La IKBa es fosforilada por IKKβ en el extremo N-ter en residuos de serina, lo que permite la unión de IkB a con la proteína que contiene repeticiones de transducción p (pTrCP), la cual forma parte del complejo de ligasa de ubiquitina Skp1/Cul1/F-box (SCF) (21). Éste cataliza la rápida poliubiquitinación de IkB a para su degradación por el proteosoma 26S, permitiendo la liberación de NF-κB y la consiguiente modulación de transcripción de genes (15).
Ruta alternativa o no canónica
La ruta alternativa, independiente de NEMO, es inducida en respuesta al factor activador de células B (BAFF) (22), LT(3, ligando CD40, virus de leucemia humana tipo I de células T (HTLV) y virus de Epstein-Barr (EBV), y en ella participa la cinasa inductora de NF-κB (NIK). NIK fosforila al homodímero IKKα desencadenando su interacción con la proteína p100 que está unida a RelB. La fosforilación de p100 es seguida por su procesamiento en proteosoma a p52 y por la translocación del complejo RelB/ p52 al núcleo (3).
Ruta inducida por luz ultravioleta (UV)
La activación de esta ruta es independiente de daño en el DNA y la degradación de IkBoc es también independiente de la activación de las IKKs. En esta ruta IkBoc es fosforilada en el C-ter por una serina/treonina, la caseína II cinasa (CKII). La CKII es activada por la p38-MAPK en respuesta a estrés por luz UV. Esta vía desempeña un papel muy importante en la protección contra luz UV mediante la inducción de la expresión de genes antiapoptóticos dependientes de NF-κB (23).
Ruta inducida en respuesta a estrés genotóxico, daño en DNA y estrés oxidativo
Los principales agentes inductores de estrés son los radicales libres de oxígeno y nitrógeno, ROS y NOR respectivamente; éstos se producen endógenamente como resultado del metabolismo proteico y metabolismo mitocondrial. Durante la respuesta inmune los eosinófilos, neutrófilos y macrófagos producen radicales libres (24).
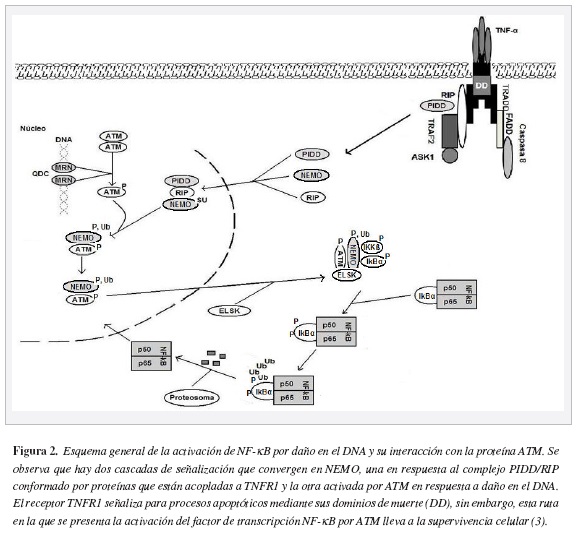
Las especies reactivas activan al NF-κB mediante un proceso en el cual NEMO desempeña un papel importante. A diferencia de la ruta clásica en la que se presenta ubiquitinación de NEMO, en la vía de señalización desencadenada por estrés oxidativo, estrés genotóxico y daño en el DNA, ocurre una modificación característica en el dominio de dedos de Zn (ZF) de NEMO, uniéndose una molécula pequeña similar a la ubiquitina (SUMO) en las Lys 277 y 309, provocando la translocación de NEMO al núcleo, sin alterar su estabilidad. Esta sumoilación de NEMO es desencadenada exclusivamente por compuestos que dañan el DNA o por quiebres de doble cadena de DNA, pero no en respuesta a citocinas (25).
En el proceso de transporte de NEMO al núcleo interviene el complejo conformado por la proteína con dominio de muerte inducida por p53 (PIDD) y la proteína interactuante de receptor (RIP). El ingreso de PIDD/RIP/NEMO al núcleo, permite la interacción de NEMO con la proteína de ataxia telangiectasia mutada (ATM). Es de anotar que la proteína ATM se activa como respuesta a modificaciones en cromatina y daño en el DNA.
En el núcleo, después de su desumoilación NEMO es fosforilado por ATM; luego de la ubiquitinación de NEMO el complejo ATM/ NEMO es translocado al citoplasma donde recluta a las IKK (a y (3) y a la ELKS. La fosforilación de la proteína IkBoc lleva a su degradación por el proteosoma permitiendo la translocación de NF-κB al núcleo (26).
La relación entre NF-κB y enzimas antioxidantes parece ser de gran importancia en la supervivencia celular. Se han encontrado elementos de unión a NF-κB en los promotores de los genes superóxido dismutasa (27), catalasa (28) y glutatión (29). Estas enzimas participan en el control de los niveles de especies reactivas de oxígeno en la célula (Figura 2).
Inhibidores de la activación de NF-κB
La disminución en el proceso de apoptosis puede conducir a cáncer y enfermedades autoin-munes, mientras que un exceso de apoptosis puede desempeñar un papel en patologías como el síndrome de inmunodeficiencia adquirida (AIDS) y enfermedades neurológicas como Alzheimer, atrofia muscular y corea de Hunting-ton (30). En esos procesos, el factor de transcripción NF-κB juega un papel muy importante ya que varios mediadores del proceso inflamatorio dependen de este factor transcrip-cional (31). El NF-κB tiene un rol potencial en enfermedades inflamatorias tales como artritis reumatoidea y asma debido a su papel inductor de citocinas proinflamatorias, quimocinas, enzimas promotoras de inflamación, inmunore-ceptores y moléculas de adhesión (32).
A20, proteína inhibidora de la activación de NF-κB
La proteína A20 es codificada por un gen que fue originalmente identificado como un gen inducible por TNF-α en células endoteliales de cordón umbilical.
En algunas líneas celulares, A20 fue inicialmen-te caracterizada como un inhibidor de la apoptosis inducida por TNF-α (33); los mecanismos que intervienen en esta inhibición no han sido totalmente aclarados, pero se sabe que están correlacionados con la inhibición de la fosfolipasa A (PLA) y la consecuente disminución de la producción de especies reactivas de oxígeno, el colapso del potencial de membrana mitocondrial y la activación de las caspasas como la caspasa 3 (34).
Además de sus propiedades antiapoptóticas, A20 puede actuar como un inhibidor de NF-κB, ya que se ha observado que la sobreexpresión de A20 bloquea la activación de NF-κB por TNFa, IL-1, LPS, ésteres de forbol y peróxido de hidrogeno en diferentes tipos celulares (35). Así mismo la sobreexpresión de A20 previene la transcripción de los genes de proteínas dependientes de NF-κB como E-selectina, molécula de adhesión-1, IkBoc, IL6, IL-8 y el factor estimulante de colonias de granulocitos-macrófagos (36). A su vez, la transcripción de A20 es regulada por NF-κB (37).
La proteína A20 se une a NEMO impidiendo la activación de IKK(3 y por ende la translocación de NF-κB al núcleo (ruta clásica). La A20 no tiene efecto en la activación de NF-κB vía p38 MAPK (35) ni NIK (38).
La poliubiquitinación de RIP es muy importante para su actividad cinasa, la polimerización de las ubiquitinas determina si será o no degradado en proteosoma; se ha encontrado que la poliubiquitinación en la Lys63 protege contra la degradación, mientras si ocurre en la Lys48 habrá degradación de las proteínas en proteosoma.
La proteína A20 induce la desubiquitinación en las Lys63 de ubiquitinas unidas a RIP. Se ha postulado que NEMO compite con A20 por la Lys 63 ubiquitinada de RIP, señal importante en la activación de TAK que permite la acción cinasa de las IKK sobre el inhibidor de NF-κB (39).
Medicamentos utilizados para inhibir la activación/actividad de NF-κB.
La alteración en la ruta alternativa de la señalización de NF-κB ha sido asociada con un amplio rango de desórdenes como la artritis reumatoidea, la colitis ulcerativa y los linfomas de células B. Los inhibidores de la ruta alternativa han sido evaluados como herramientas terapéuticas en estas patologías (40).
Las alteraciones en la vía de señalización de NF-κB han sido asociadas con enfermedades inflamatorias y cáncer (40). Se han descrito los efectos sobre esta ruta de fármacos antiinflamatorios e inmunosupresores y se han venido desarrollando medicamentos de tecnología molecular que tienen como objetivo terapéutico modificar la vía de señalización de NF-κB (41). Además, se ha explorado la posibilidad de combinar terapias convencionales con tratamientos derivados de la biotecnología en el manejo de las patologías que involucran las rutas de NFκB (42).
Medicamentos anti-inflamatorios no esteroides (NSAID)
Cada vez hay más estudios que asocian la inflamación al cáncer. Investigaciones epidemiológicas han mostrado que la ingesta de ácido acetil salicílico (ASA) ejerce un efecto protector contra la aparición del cáncer colorectal. Además se ha demostrado que el ASA es un quimioprotector (43). El ASA inhibe la síntesis de prostaglandinas a través de su acción sobre la actividad de la ciclooxigenasa (COX) (44). Varios estudios sugieren que el efecto proa-poptótico de NSAID es debido a la inhibición de NF-κB, la cual ocurre a diferentes niveles: actividad de las cinasas IKK, inhibición de la degradación de los IκBs, translocación nuclear de NF-κB, unión al DNA y actividad transcripcional del complejo NF-κB (45).
La inhibición de IKK por el ASA es resultado de la competencia por el sitio de unión del adenosin trifosfato (ATP) en la IKK(3 (46). Los efectos de los NSAID en la actividad de NF-κB dependen de las líneas celulares y son específicos de medicamento. La indometacina y el ibuprofeno son más efectivos que el ASA en inhibir la activación de NF-κB vía TNF-a en líneas celulares de leucemia (47) mientras que en líneas de cáncer de colon no tienen efecto sobre la vía de NF-κB (48 46). El ibuprofeno inhibe la activación constitutiva de IKKα en tumores de próstata independientes de andrógenos (44).
Recientemente se ha observado en líneas celulares de cáncer de colon, en ausencia de citocinas adicionales, que el efecto a largo plazo del ASA está mediado por la regulación de la degradación de IkBoc y la relocalización de RelA en el nucléolo la cual inhibe la actividad transcipcional de NF-κB y favorece la apoptosis (49). Es de anotar que la relocalización en nucléolo inhibe la actividad transcripcional de RelA (50).
Varios estudios indican que el ASA y otros NSAID presentan una actividad quimio-protectora contra varios tipos de cáncer como el de colon, pulmón, linfoma de Hodking, esófago, próstata, y contra otros en los que la vía de NF-κB es importante (51).
Glucocorticoides
Los glucocorticoides (GCs), que son ampliamente utilizados como anti-inflamatorios e inmunosupresores, ejercen sus funciones a través de sus receptores (GR), provocando efectos inhibidores sobre la transcripción desencadenada por NF-κB (52).
Se han reportado varios mecanismos mediante los cuales los GR activados inhiben la vía de señalización de NF-κB. Así por ejemplo, en linfocitos y monocitos se ha demostrado que la dexametasona induce un incremento en la transcripción del gen de IκBα favoreciendo el secuestro de NF-κB en citoplasma. Sin embargo este efecto no se observa en células endoteliales (53).
También se ha observado que la disminución de la actividad transcripcional de NF-κB en respuesta a los GCs es debida a una interacción del GR activado con RelA, conduciendo a la liberación de los factores coactivadores y a un reclutamiento de factores correpresores como la deacetilasa C2 de histonas (HDAC2). Esta interacción también podría causar una disminución de la fosforilación de la RNA polimerasa II, disminuyendo así la actividad transcripcional. Se han demostrado estos efectos de represión del GR en los promotores de los genes de IL-8 e ICAM-1 (54).
Moduladores selectivos de receptor de estrógenos (SERMs)
De manera similar a lo que ocurre con los GCs, los SERMs pueden modular la actividad de NF-κB. Estudios preliminares han demostraron que el tamoxifeno reduce la activación de NF-κB vía TNF-α en ciertas líneas celulares. Este efecto es mediado a través de la inhibición de la actividad de las cinasas IKK y la supresión de la degradación de IκBα (47).
Otros estudios han demostrado que el raloxifeno y el tamoxifeno son inhibidores de la actividad de NF-κB en líneas celulares de mieloma; se ha sugerido que el raloxifeno podría bloquear la actividad constitutiva de NF-κB a través de la modulación de la asociación del receptor de estrógenos (ER) con la proteína RelA. El raloxifeno y el tamoxifeno inducen apoptosis e incrementan la respuesta a agentes citotóxicos como vincristina y trióxido arsénico en el tratamiento del mieloma múltiple (44).
Ligandos del receptor activado por proliferadores de peroxisoma (PPARs)
La regulación de NF-κB por agonistas de PPARy podría ser una estrategia para el tratamiento del cáncer. Se ha demostrado que la tiazolidinediona presenta un efecto proapoptótico dependiente de NF-κB en enfermedades malignas de células B y en cáncer de colon. A su vez, se ha demostrado que el efecto proapoptótico de la 15 desoxy-prostaglandina J2 (15 desoxy-PGJ2) (un ligando endógeno de PPARy) y la ciglitazona están relacionados con la sobreexpresión de IkBoc e IxBp y por lo tanto con la inhibición constitutiva de la actividad de NF-κB (55).
Talidomida y análogos inmunomoduladores (IMiDs)
La talidomida y sus análogos, la lenalidomida y el actimid (también conocido como CC-4047), son medicamentos inmunomoduladores (IMiDs) que inhiben la activación de NF-κB en la vía de señalización de TNF-a e IL-1p por medio de un mecanismo que involucra la supresión de IKK y consecutivamente provocan la reducción de la expresión de moléculas proangiogénicas y antiapoptóticas, tales como IL-8 y IAP2 (56). Los IMiDs presentan efectos antiinflamatorios, antiangiogénicos, antiproliferativos y proapop-tóticos (57). Los IMiDs se utilizan en el tratamiento del eritema nodoso por lepra, mieloma múltiple y síndromes mielodisplásicos; también han demostrado utilidad en el manejo de otras patologías hematológicas, tumores sólidos y enfermedades inflamatorias (58,59). La lenalidomida y el actimid son más potentes que la talidomida y presentan menos efectos neurotóxicos (58).
Otras moléculas cuya expresión disminuye en respuesta a IMiDs son c-IAP2 y FLIP; la desregulación de estos inhibidores de caspasas en células de mieloma múltiple puede contribuir a potenciar la susceptibilidad a la apoptosis vía FAS y TRAIL (60).
Inhibidores del sistema ubiquitin-proteosoma
El bortezomib (llamado también velcade) fue descrito recientemente como un potente agente citotóxico (61). El bortezomib es un inhibidor del proteosoma 20S, el cual constituye, en conjunto con dos partículas 19S, el complejo multicatalítico 26S. El proteosoma 20S es la estructura catalítica del complejo, mientras que las partículas 19S contienen los sitios de unión para las cadenas de ubiquitina. El bortezomib evita por ende la degradación de IκBα impidiendo, por lo tanto, la activación de NF-κB (62); es un fármaco que se emplea en el tratamiento del mieloma múltiple y que podría ser útil en el manejo de otros cánceres hematológicos y tumores sólidos (44,63).
Otro compuesto que podría llegar a tener uso en la terapia de enfermedades que involucran la vía de señalización de NF-κB es un derivado del ácido benzoico llamado GS143. El GS143 inhibe la ubiquitinación de IκBα al impedir la interacción de la forma fosforilada de IκBα con el complejo de ligasa de ubiquitina SCF (64).
Medicamentos de tecnología molecular
El silenciamiento o desregulación de la expresión genética ha mostrado ser promisorio en el tratamiento de enfermedades inflamatorias y cáncer teniendo como blancos los oncogenes y genes involucrados en inflamación, angiogénesis, metástasis, supervivencia, antiapoptosis y resistencia a quimioterapia (65). La técnica de RNA de interferencia (siRNA) y la de los oligo-nucleótidos de DNA de doble cadena con elementos cis podrían ser herramientas terapéuticas en patologías que comprometen la vía de señalización de NFkB (44,66).
Se ha demostrado en la línea de células tumorales humanas de carcinoma colorectal HCT116 que el p65 siRNA bloquea la expresión de p65 y aumenta la sensibilidad celular a los efectos citotóxicos del irinotecan (CPT-11), un inhibidor de la topoisomerasa I. A su vez, en ratones atímicos desnudos inyectados por vía subcutánea con células HCT116, se ha observado una mayor sensibilidad al CPT-I si estos tumores han sido previamente tratados con siRNA (67). Por lo tanto, se concluye que la transfección de siRNA dirigido contra p65 aumenta, en este modelo, la susceptibilidad a la quimioterapia (42).
La tecnología de los oligonucleótidos de doble cadena con elementos cis (señuelos) se emplea para evitar que un factor transcripcional se una a su secuencia blanco en el DNA. Se ha demostrado en un modelo murino que la transfección de señuelos contra NF-κB del adenocarcinoma de colon inhibe la caquexia, sugiriendo la posible utilidad de esta tecnología en el manejo de la caquexia asociada al cáncer (68). Además, la transfección de señuelos contra NF-κB del reticulosarcoma M5076 disminuye en el ratón las metástasis hepáticas, dando muy buenos resultados cuando se combina con un medicamento anticanceroso, la mitomicina C (69).
Por otra parte, la transfección de señuelos contra NF-κB mediante administración intraar-ticular en un modelo de rata con artritis inducida por colágeno disminuye la inflamación, la destrucción articular y la producción sinovial de IL-1 y TNF-a (70). A su vez, in vitro, la transfección de señuelos contra NF-κB produce inhibición de la producción de IL-1β, IL-6, TNF-a, ICAM-1 y metaloproteinasa-1 de la matriz en células sinoviales de pacientes con artritis reumatoide (71).
Conclusiones
Las moléculas más importantes del signalosoma de NF-κB son las IKKα, IKK(3 y NEMO en la ruta canónica, IKKα y NIK en la no canónica, MAPK (p38) por luz ultravioleta y ATM, PIDD/ RIP/NEMO, IKKα e IKK(3 en la de estrés genotóxico, estrés oxidativo y daño en el DNA. Según la vía ocurre la formación de homodímeros o heterodímeros de la familia Rel lo que regula la expresión de diferentes genes. El tiempo de activación de NF-κB varía según la vía de señalización, siendo más rápida para la canónica (algunos minutos) y más lenta para estrés genotóxico (aproximadamente 90 minutos); con efecto más duradero en esta última (25).
En los procesos de apoptosis y supervivencia de la célula vía TNFR se encuentran c-Jun N-ter cinasa (JNK) que generalmente induce procesos de muerte y el NF-κB que potencia procesos de supervivencia inhibiendo a JNK.
NF-κB disminuye la acumulación de ROS en respuesta a TNF-a, lo que es muy importante ya que ROS son mediadores de apoptosis inducida por JNK y son requeridas para la activación sostenida de ésta. Así, NF-κB ésta relacionado con procesos antioxidantes y posterior inactivación de JNK (72). Se podría plantear que la capacidad antioxidante de una célula es determinante en el curso hacia la ruta de apoptosis o supervivencia.
Varios estudios demuestran que la resistencia a radio y quimioterapia está asociada con un incremento de la actividad de enzimas antioxidantes. El uso de terapias concomitantes con estos inhibidores del NF-κB podría sensibilizar la célula a la muerte programada.
Referencias
1. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappa B by a posttranslational mechanism. Cell. 1986;47:921-928.
2. Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195-2224.
3 . Gloire G , Legrand-Poels S , Piette J . NF-κB activation by reactive oxygen species : Fifteen years later . Biochem Pharmacol . 2006 ; 72 : 1493-1505 .
4. Beinke S, Ley S. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J. 2004;382:393-409.
5. Fujita T, Nolan G, Liou H, Scott M, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through p50 homodimers. Genes Dev. 1993;7:1354-1363.
6. Farrow B, Evers B. Inflammation and the develop-ment of pancreatic cancer. Surg Oncol. 2002;10:153 169.
7. Gerondakis S, Morrice N, Richardson I, Wetten-hall R, Fecondo J, Grumont R. The activity of a 70kDa IκB molecule identical to the carboxyl terminus of the p105 NF-κB precursor is modulated by protein kinase A. Cell Growth Differ. 1993;4:617-627.
8. Chen L, Greene W. Shaping the nuclear action of NF-κB. Nat Immunol Rev. 2004;5:392-401.
9. Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231-1240.
10. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91: 243-252.
11. Lee JI, Burckart GJ. Nuclear factor kB: Important transcription factor and therapeutic target. J Clin Pharmacol. 1998;38:981-993.
12. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18:621-663.
13. May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-ka-ppaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550-1554.
14. Verma UN, Yamamoto Y, Prajapati S, Gaynor RB. Nuclear role of I kappa B kinase-gamma/NF-kappa B essential modulator (IKK gamma/NEMO) in NF-kappa B-dependent gene expression. J Biol Chem. 2004;279:3509-3515.
15. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280-288.
16. Martin A, Fresno M. Tumor necrosis factoralpha activation of NF-κB requires the phosphorylation of Ser-471 in the transactivation domain of c-Rel. J Biol Chem. 2000;275:2438324391.
17. O'Neill LA. How Toll-like receptors signal: what we know and what we don't know. Curr Opin Immunol. 2006;18:3-9.
18. Weil R, Israel A. T-cell-receptor- and B-cell-receptor-mediated activation of NF-kappaB in lymphocytes. Curr Opin Immunol. 2004;16:374-381.
19. Pomerantz J, Baltimore D. Two pathways to NF-κB. Mol Cell. 2002;10:693-701.
20. Takaesu G, Surabhi R, Park K, Ninomiya-Tsuji J, Matsumoto K, Gaynor R. TAK1 is critical for IκB kinase-mediated activation of the NF-κB pathway. J Mol Biol. 2003;326:105-115.
21. Liu Y. Ubiquitin ligases and the immune response. Annu Rev Immunol. 2004; 22:81-127.
22. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958-965.
23. Kato T, Delhase M, Hoffmann A, Karin M. CKII is a C-terminal IκB kinase responsible for NF-κB activation during UV response. Mol Cell. 2003;12:829-839.
24. Conner E, Grisham M. Inflammation, free radicals, and antioxidants. Nutrition. 1996;12:274-277.
25. Huang T, Wuerzberger-Davis S, Wu Z, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565-576.
26. Habraken Y, Piette J and Piret B. S phase dependence and involvement of NF-kappaB activating kinase to NF-kappaB activation by camptothecin. Biochem Pharmacol. 2001;62:603-616.
27. Guo G, Yan-Sanders Y, Lyn-Cook B, Wang T, Ta-mae D, Oqi J, et al. Manganese Superoxide Dismu-tase-Mediated Gene Expression in Radiation-Induced Adaptive Responses. Mol Cell Biol. 2003;23:2362-2378.
28. Zhou L, Johnson A, Rando T. NF-κB and AP-1 mediate transcriptional responses to oxidative stress in skele-tal muscle cells. Free Radic Biol Med. 2001; 31:1405-1416.
29. Morales A, Miranda M, Sanchez-Reyes A, Colell A, Biete A, Fernández-Checa J. Transcriptional regulation of the heavy subunit chain of g-glutamylcysteine synthetase by ionizing radiation. FEBS Letters. 1998; 427:15-20.
30. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456-1462.
31. Grossmann M, Nakamura Y, Grumont R, Gerondakis S. New insights into the roles of ReL/NF-κB transcription factors in immune function, hemopoiesis and human disease. Int J Biochem Cell Biol. 1999;31:1209-1219.
32. Meager A. Cytokine regulation of cellular adhesion molecule expression in inflammation. Cytokine Growth Factor Rev. 1999;10:27-39.
33. Opipari AW Jr, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992;267:12424-12427.
34. Wissing D, Mouritzen H, Jaattela M. TNF-induced mitochondrial changes and activation of apoptotic proteases are inhibited by A20. Free Radic Biol Med. 1998;25:57-65.
35. Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000; 2: 301-311.
36. Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, Bach FH. A20 inhibits NF-κB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apopto-sis. Blood. 1998;91:2249-2258.
37. Beyaert R, Heyninck K, Van Huffel S. A20 and A20-Binding proteins as cellular inhibitors of nuclear factor-κB-dependent gene expression and apoptosis. Biochem Pharmacol. 2000;60:1143-1151.
38. Heyninck K, De Valck D, Vanden Berghe W, Van Criekinge W, Contreras R, Fiers W, et al. The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB inhibiting protein ABIN. J Cell Biol. 1999;145:1471-1482.
39. Chen F, Bhatia D, Chang Q and Castranova V. Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ. 2006;13:1835-1838.
40. Sun X, Zhang H.NFKB and NFKBI polymorphisms in relation to susceptibility of tumour and other diseases. Histol Histopathol. 2007;22:1387-1398.
41. Dejardin E. The alternative NF-κB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161-1179.
42. Veiby OP, Read MA. Chemoresistance: Impact of nuclear factor (NF)-κB inhibition by small interfering RNA. Clin Cancer Res. 2004;10:3333-3341.
43. Chan A, Giovannucci E, Schernhammer E, Coldi-tz G, Hunter D, Willett W, et al. A prospective study of aspirin use and the risk for colorectal adenoma. Ann Intern Med. 2004;140:157-166.
44. Olivier S, Robe P, Bours V. Can NF-κB be a target for novel and efficient anti-cancer agents? Biochem Pharmacol. 2006;72:1054-1068.
45. Kopp E, Ghosh S. Inhibition of NF-kappa B by so-dium salicylate and aspirin. Science. 1994;265:956-959.
46. Yin M, Yamamoto Y, Gaynor R. The anti-inflamma-tory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77-80.
47. Takada Y, Bhardwaj A, Potdar P, Aggarwal BB. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene. 2004;23:9247-9258.
48. Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulin-dac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999;274:27307-27314.
49. Stark L, Dunlop M. Nucleolar sequestration of RelA (p65) regulates NF-kappaB-driven transcription and apoptosis. Mol Cell Biol. 2005;25:59856004.
50. Ito K, Barnes P, Adcock I. Glucocorticoid receptor recruitment of HDAC 2 inhibits interleukin-1-β-induced histone H4 acetilation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891-6903.
51. Chang E, Zheng T, Weir E, Borowitz M, Mann R, Spiegelman D, et al. Aspirin and the risk of Hodgkin's lymphoma in a population-based casecontrol study. J Natl Cancer Inst. 2004;96:305-315.
52. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488-522.
53. Scheinman RI, Cogswell PC, Lofquist AK, Bald-win Jr AS. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283-286.
54. Kagoshima M, Wilcke T, Ito K, Tsaprouni L, Bar-nes PJ, Punchard N, et al. Glucocorticoid-mediated transrepression is regulated by histone acetylation and DNA methylation. Eur J Pharmacol. 2001;429:327-334.
55. Ray DM, Akbiyik F, Bernstein SH and Phipps RP. CD40 engagement prevents peroxisome proliferatoractivated receptor gamma agonist-induced apoptosis of B lymphocytes and B lymphoma cells by an NF-kappaBdependent mechanism. J Immunol. 2005;174:4060-4069.
56. Keifer JA, Guttridge DC, Ashburner BP, Baldwin Jr AS. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J Biol Chem. 2001;276:22382-22387.
57. Armoiry X, Aulagner G, Facon T. Lenalidomide in the treatment of multiple myeloma: a review. J Clin Pharm Ther. 2008;33:219-226.
58. Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol. 2007; 39:1489-1499.
59. Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4:314322.
60. Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, et al. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002;99:4525-4530.
61. Adams J, Palombella VJ, Elliott PJ. Proteasome inhibition: a new strategy in cancer treatment. Invest New Drugs. 2000;18:109-121.
62. Adams J, Palombella VJ, Sausville EA, Johnson J, Descree A, Lazarus DD et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615-2622.
63. Richardson P, Mitsiades C, Hideshima T. and Anderson C. Bortezomib: proteasome inhibition as an effective anticancer therapy, Annu Rev Med. 2006;57:33-47.
64. Nakajima H, Fujiwara H, Furuichi Y, Tanaka Kei-ji, Shimbara N. A novel small-molecule inhibitor of NF-κB signaling. Biochem Biophys Res Comun. 2008; 368:1007-1013.
65. Bhindi R, Fahmy RG, Lowe HC, Chesterman CN, Dass CR, Cairos MJ, et al. Brothers in arms: DNA enzymes, short interfering RNA and the emerging wave of small-molecule nucleic acid-based gene-silencing stra-tegies. Am J Pathol. 2007;171:1079-1088.
66. Morishita R, Tomita N, Kaneda Y, Ogihara T. Molecular therapy to inhibit NFkappaB activation by transcription factor decoy oligonucleotides. Curr Opin Pharmacol. 2004;4:139-146.
67. Guo J, Verma U, Gaynor R, Frenkel E, Becerra C. Enhanced chemosensitivity to irinotecan by RNA interference. Mediated down-regulation of the nuclear factor-κB p65 subunit. Clinical Cancer Res. 2004;10:3333-3341.
68. Kawamura I, Morishita R, Tomita N, Lacey E, Aketa M, Tsujimoto S, et al. Intratumoral injection of oligonucleotides to the NFkB binding site inhibits cachexia in a mouse tumor model. Gene Ther. 1999;6:91-97.
69. Kawamura I, Morishita R, Tsujimoto S, Manda T, Tomoi M, Tomita N, et al. Intravenous injection of oligodeoxynucleotides to the NF-kappaB binding site inhibits hepatic metastasis of M5076 reticulosarcoma in mice. Gene Ther. 2001;8:905-912.
70. Tomita T, Takeuchi E, Tomita N, Morishita R, Kaneko M, Yamamoto K, et al. Suppressed severity of collagen-induced arthritis by in vivo transfection of nuclear factor kappaB decoy oligodeoxynucleotides as a gene therapy. Arthritis Rheum. 1999;42:2532-2542.
71. Tomita T, Takano H, Tomita N, Morishita R, Ka-neko M, Shi K, et al. Transcription factor decoy for nuclear factor kappaB inhibits cytokine and adhesion molecule expressions in synovial cells derived from rheumatoid arthritis. Rheumatology. 2000;39:749-757.
72. Pham C, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, et al. Ferritin heavy chain upregulation by NF-κB inhibits TNF-α-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529-542.
73. Haddad J. Oxygen sensing and oxidant/redox-related pathways. Biochem Biophys Res Commun. 2004;316:969-977.
74. Xu Y, Porntadavity S and Clair D. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2). Biochem J. 2002;362:401-412.