review article
DOI: https://doi.org/10.15446/revfacmed.v67n3.70149
Epstein-Barr virus infection as a predisposing factor for multiple sclerosis. An update from molecular biology, immunology and epidemiology
Infección por virus de Epstein-Barr como factor predisponente en la esclerosis múltiple. Actualización desde la biología molecular, la inmunología y la epidemiología
Received: 31/01/2018. Accepted: 25/06/2018.
David López-Valencia1,3 • Ángela Medina-Ortega1,3 • Diego Fernando Hoyos-Samboní2 • Jhan Sebastián Saavedra-Torres1,3 Carolina Salguero3
1 Universidad del Cauca - Faculty of Health Sciences - Popayán - Colombia.
2 Empresa Social del Estado Hospital San Vicente de Paul Mistrató - Emergency Service - Mistrató, Risaralda - Colombia.
3 Del Lab al Campo - BIOTECMED Research Seedbed - Bogotá D.C. - Colombia.
Corresponding author: David López-Valencia. Facultad de Ciencias de la Salud, Universidad del Cauca. Carrera 6 No. 13N-50. Phone number: +57 2 8234118. Popayán. Colombia. Email: davlopez@unicauca.edu.co.
| Abstract |
Introduction: Epstein-Barr virus is an infectious agent used to immortalize and induce polyclonal activation of B cells. It has been widely described that this virus produces changes in the cells it infects and in the immune response, and stimulates the development of autoimmune diseases.
Objective: To characterize the association between Epstein-Barr virus and multiple sclerosis described in current scientific literature.
Materials and methods: A 59-years range literature search was conducted in the PubMed, ScienceDirect, Redalyc and SciELO databases using the following MeSH terms: “Epstein-Barr virus, multiple sclerosis autoimmune diseases, autoimmune diseases of the nervous system”.
Results: Many studies describe the association between Epstein-Barr virus and multiple sclerosis. It is believed that acute infection and viral reactivation promote the development of multiple sclerosis.
Conclusions: It is necessary to conduct further research on the pathogenesis and morphophysiological and neuroimmunological changes –at the ecological, molecular, cellular, tissue, organic and systemic level– induced by the immune response and that favor the development of multiple sclerosis.
Keywords: Epstein-Barr Virus; Multiple Sclerosis; Relapse; Autoimmune Diseases of the Nervous System (MeSH).
López-Valencia D, Medina-Ortega A, Hoyos-Samboní DF, Saavedra-Torres JS, Salguero C. Epstein-Barr virus infection as a predisposing factor for multiple sclerosis. An update from molecular biology, immunology and epidemiology. Rev. Fac. Med. 2019;67(3):493-501. English. doi:
https://doi.org/10.15446/revfacmed.v67n3.70149.
| Resumen |
Introducción. El virus de Epstein-Barr (VEB) es un agente inmortalizador y activador policlonal de células B. Es conocido que este virus induce cambios en las células que infecta y en la respuesta inmune, y que favorece la presentación de enfermedades autoinmunes.
Objetivo. Caracterizar la asociación entre el VEB y la esclerosis múltiple (EM) descrita en la literatura actual.
Materiales y métodos. Se realizó una búsqueda bibliográfica con rango de 59 años mediante los términos DeCS “virus de Epstein-Barr, esclerosis múltiple, enfermedades autoinmunes del sistema nervioso” en las bases de datos PubMed, ScienceDirect, Redalyc y SciELO.
Resultados. Hay muchos estudios que describen la asociación del VEB con la EM. Se cree que la infección aguda y la reactivación viral contribuyen al desarrollo de la enfermedad.
Conclusiones. Es necesario realizar más estudios que indaguen sobre la patogénesis, los cambios morfofisiológicos y las alteraciones neuroinmunológicas en la ecología molecular, celular, tisular, orgánica y sistémica inducida por la respuesta inmune y que favorecen el desarrollo de la EM.
Palabras clave: Virus de Epstein-Barr; Esclerosis múltiple; Enfermedades autoinmunes del sistema nervioso (DeCS).
López-Valencia D, Medina-Ortega A, Hoyos-Samboní DF, Saavedra-Torres JS, Salguero C. [Infección por virus de Epstein-Barr como factor predisponente en la esclerosis múltiple. Actualización desde la biología molecular, la inmunología y la epidemiología]. Rev. Fac. Med. 2019;67(3): 493-501. English. doi: https://doi.org/10.15446/revfacmed.v67n3.70149.
Introduction
The Epstein-Barr virus (EBV) was discovered in 1964 by the British pathologist Sir Michael A. Epstein, who suggested that it played a major role in the pathogenesis of Burkitt lymphoma. Epstein was able to publish this claim after observing electro microscopy images of EBV-infected lymphoblast cell cultures taken by Yvonne Barr and Bert Achong. (1,2) Since then, several studies have investigated the immune response against EBV in autoimmune diseases, which has never been associated with a clearly defined cause.
EBV is an immortalizing agent and polyclonal activator of B cells (3) that induces modifications in plasma membranes and leads to immunomodulation imbalance between regulatory T cells (Tregs), Th1/Th2/Th3/Th17 responses, cytotoxic T cells (CD8+), NK cells and the signaling pathways that control cell viability and survival. All these changes favor the development of autoimmune diseases, such as multiple sclerosis (MS), in genetically predisposed individuals. (4-6)
MS is a chronic inflammatory disorder of the central nervous system that may have lead to the development of optic neuritis, brain-stem dysfunction, or transverse myelitis at some point. (7) In recent decades, several researchers have studied the possible association of EBV with autoimmune disorders, and, even though this association seemed unrealistic at first, and the results did not completely convince the medical community, more compelling results on the correlation between EBV and MS have been published recently. However, as of today, no direct causality has yet been determined.
The objective of this article is to review the most recent literature on the EBV-MS association and to propose several alternatives to improve the management of this condition in Colombia.
Materials and methods
A structured bibliographic search was performed with a range of 59 years (1960-2019) in order to collect historical and updated information on the EBV-MS association. The search was made in English and Spanish in the following databases: PubMed, ScienceDirect, Redalyc and SciELO. The following MeSH terms were used: “Epstein-Barr virus, multiple sclerosis, autoimmune diseases of the nervous system”, and the equivalent DeCS terms in Spanish: “Virus de Epstein-Barr, esclerosis múltiple, enfermedades autoinmunes del sistema nervioso”. The combination of operators used in English was “Epstein-Barr virus AND multiple sclerosis” and “Epstein-Barr virus AND autoimmune diseases of the nervous system”, and in Spanish “Virus de Epstein-Barr Y enfermedades autoinmunes del sistema nervioso” and “Virus de Epstein-Barr Y enfermedades autoinmunes del sistema nervioso”.
The following types of study were included: original research, review articles, opinion articles, case reports, reports, and books focusing on historical, biological, immunological, epidemiological and experimental aspects of the findings of the EBV-MS association. Books with relevant information about the association were also reviewed.
In total, 8 674 references were collected, of which 8 594 were excluded (7 892 because access to their full text was not available, 645 because they were not relevant to this article, and 57 because they were duplicates); thus, 80 references were finally included for analysis (Figure 1). The open access software Mendeley version 1.18 was used to manage and organize the information. The theoretical framework reflected in the structure of the article was built by analyzing results, discussions and conclusions using the following classification: EBV epidemiology, EBV molecular biology, EBV and autoimmune diseases, MS, MS epidemiology, genetic risk factors in MS, and EBV-MS association.

Figure 1. Bibliographic search flowchart.
Source: Own elaboration.
Results
A large number of studies describing the EBV-MS association was found. After removing duplicates, 80 articles were finally selected for inclusion based on their relevance and full text availability. No emphasis was placed on particular populations or regions. These articles were classified as follows; 40 original investigations, 26 review articles, 4 meta-analyses, 4 opinion articles, 4 case reports, 1 report and 1 book. Table 1 lists some of the most significant references that were included.
Table 1. Most significant references for this article.
|
Authors |
Country/region/population studied |
Language |
Year |
Key findings |
|
Ascherio et al. (8) |
U.S.A. 62 439 women who gave blood samples between 1989-1990 and 1996-1999. |
English |
2001 |
Compared with matched controls, women with MS had higher geometric mean anti-EBV antibody titers, but not for cytomegalovirus. The increases were significant for anti-EBNA-1 (GMT, 515 vs. 203, p=0.03), anti-EBNA-2 (GMT, 91 vs. 40, p=0.01), and anti-EA-D antibodies (15.9 vs. 5.9, p=0.04). A difference >4 times was associated with a relative risk of MS of 3.9 (95%CI: 1.1-13.7). The results support a role the EBV in the etiology of MS. |
|
Kakalacheva et al. (9) |
Switzerland 35 IM patients and 23 control subjects. |
English |
2016 |
They detected frequent reactivity of anti-vimentin IgM in 27 (77%) patients with IM, but only in 2 of the control subjects (9%). IgG response to myelin oligodendrocyte glycoprotein (MOG) was observed in 40% of children with central nervous system demyelinating inflammatory diseases. They were detectable in 7 (20%) IM patients, but not in control subjects. Anti-vimentine IgM and anti-MOG IgG response decreased following clinical resolution of acute IM symptoms. MOG-specific memory B cells are activated in a subgroup of IM patients. |
|
Casiraghi et al. (10) |
Canada. 3 MS patients who donated brain microvascular endothelial cells (autopsy). |
English |
2011 |
They demonstrated satisfactory EBV infection and gene expression in brain microvascular endothelial cells. EBV infection could activate and increase the production of molecules involved in the adhesion of leukocytes to the endothelium (CCL-5, ICAM-1). These molecules have been associated with MS; polymorphisms in CCL-5 and their CCR5 receptor modify the course and outcome of MS. An increased number of peripheral blood mononuclear cells adhered to brain microvascular endothelial cells (p<0.0001). Brain endothelial cells may regulate high cytokines, chemokines, and adhesion molecules that induce a breach in the blood-brain barrier and attract lymphocytes to the brain. |
|
Zhou et al. (11) |
Australia and U.S.A. 3 599 individuals. |
English |
2016 |
They identified a locus showing strong association in the human leukocyte antigen (HLA) region, in which the most significantly associated single nucleotide polymorphism was rs2516049. (p=4.11 x 10-9). They showed that the genetic risk for elevated anti-EBNA-1 titers is positively correlated with the development of MS. |
MS: multiple sclerosis, EBV: Epstein-Barr virus, IM: infectious mononucleosis.
Source: Own elaboration.
Discussion
The EBV-MS association was approached based on what was found in the literature review search: EBV epidemiology, EBV molecular biology, EBV and autoimmune diseases, MS, epidemiology of MS and genetic risk factors for MS and EBV-MS association.
EBV epidemiology
Most primary EBV infections occur during childhood or early adolescence, but its course is usually subclinical. High seroprevalence rates have been reported in USA: Dowd et al. (12) reported a rate of 66.5% in children and young adults aged 6-19 years. If the infection occurs in late adolescence or early adulthood, it usually presents as infectious mononucleosis, since 90-95% of adults and seniors have antibodies against EBV. (13)
The most frequent mode of EBV transmission is through bodily fluids, for this reason it is commonly known as the “kissing disease”. To a lesser extent, EBV is transmitted by oral contact with sweets, food, toys, or other contaminated objects. Additionally, it is also transmitted through sexual contact, blood transfusions, hematopoietic stem cells transplantation, solid organ transplants, and during childbirth. (12,13) More than 90% of infected people sporadically release EBV throughout their lives, even if they remain asymptomatic. (4)
Molecular biology of EBV
The EBV genome is a double-stranded DNA with the ability to integrate into the genome of the host. This virus belongs to the Herpesviridae family, Gammaherpesvirinae subfamily, Lymphocryptoviruses genus, and Human Gammaherpesvirus 4 species. In 2015, the International Committee on Virus Taxonomy (ICTV) renamed it Human Gammaherpesvirus 4 (14), although it is still known as EBV in the medical jargon.
After entering the oropharyngeal epithelium, EBV can infect different cell lines such as B cells, T cells, NK cells, and epithelial cells. EBV is able to immortalize infected cells in cases where immunity is altered (HIV/AIDS, organ transplantation, prolonged steroid therapy, depression, etc.) This inmortalizing effect occurs after more than 100 EB viral gens have been integrated, since the viral proteins expression patterns vary according to the conditions of the host and the type of infection. (13,15) EBV encodes proteins that facilitate its entry to B cells: the CR2 viral receptor (CD21 binds to the C3d component of the complement) and GP350, the main viral protein that binds it to B cells through the major histocompatibility class II complex.
EBV also encodes EBNA (nuclear antigens) and LMP (latent membrane proteins), among other transcripts, that make way for it in the host. (16-18) The expression of different viral genes may vary according to the evolution of the early, latent (chronic) and transformation (neoplasm) infection (Table 2).
Table 2. Transcripts and proteins involved in the Epstein Barr virus infection cycle
|
Molecular type |
Nomenclature |
Description |
|
Nuclear antigens |
EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, EBNA-LP (Transcriptional activators of several genes) |
Expression according to the latency state. Immune evasion. Inhibition of apoptosis and immortalization of B cells by mechanisms such as inhibition of cyclin-dependent kinases and induction of Bim. |
|
Small EBV noncoding RNAs |
EBER-1, -2 (EBV nuclear antigen type 1 and 2) |
More abundant viral transcripts in cells with latent infection. Stimulation of B cells proliferation by activating cell survival pathways. Avoidance of apoptosis by stimulating IL-10 production. Induction of autocrine growth factors. EBERs accumulate in the nucleus —approximately 1 million copies per cell—, and form stable complexes with ribonucleoproteins (RNP). |
|
Micro-RNA (miRNAs) |
BHRF1, BART (EBV proteins expressed in the disease latency and cycle) |
Viral and host gene regulation. EBV produces more than 50 miRNAs that are grouped into two opposing regions of the viral genome: BamHI (BHRF1) and BamHI A (BART). |
|
Exonic sequence |
EBNA IRES |
Untranslated region of the EBNA-1 gene, which is the only nuclear protein expressed by EBV in all latencies and the lytic cycle. EBNA-1 is necessary to support the viral episome (extracromosomal replicating unit). EBNA IRES can increase EBNA1 expression by 4–14-fold in Burkitt lymphoma cells. |
|
Small nucleolar RNA |
v-snoRNA |
It is encoded by an intronic sequence within the BART region. Coding of viral DNA polymerase. Participation during viral lytic reactivation. Regulation of BALF5 (replication factor, DNA polymerase of the nucleus). |
|
Transmembrane proteins |
GP350 (Cell adhesion and penetration through CD21) |
EBV enters B cells through the interaction of the viral glycoprotein gp350 with the complement cell receptor for CD21, creating infection. When EBV binds to CD21-gp350/220, it enters by endocytosis. |
|
GP42 |
Cell adhesion and penetration by means of HLA class II. |
|
|
BMRF-2 |
Cell adhesion and penetration through integrins β1. |
|
|
GP110 (BALF-4) |
Host cell infection support. |
|
|
Glycoproteins |
gN, GP150, BDLF-2, BILF-1, BILF-2 |
Coat protein coding. |
|
Transcription factors |
BZLF-1, BRLF-1 |
Activation of the lytic replication cycle. |
|
Viral promoters |
EA, BSLF-1, BBLF-4, BBLF-2/-3, BALF-5, BALF-2 |
|
|
Viral cytokines |
IL-10 viral, BARF-1, BNLF-2A, BMLF-1, BSLF-2 |
Inhibition of interferon synthesis -γ (IFNγ). Suppression of cytotoxic activity of CTCD8+ and HLA class I expression. |
|
Restricted early antigen |
EA/R |
Immune evasion. Inhibition of apoptosis. Modulation of other host antiviral responses. |
|
Bcl-2 antagonist protein |
BALF-1 |
|
|
Latent membrane proteins |
LMP-1 |
Survival of B cells. Production of B-cell activating factor. Activation of p38, NF-κβ and JNK signaling pathway. Functional homologue of CD40, a normal B-cell protein that binds to CTCD4+ by means of CD40L in germ centers (germ center model). |
|
LMP-2A |
Survival of B cells. Functional counterpart of the B-cell receptor (BCR). |
|
|
LMP-2B |
Survival of B cells. It promotes the passage from latent infection to lytic infection. |
EBV: Epstein-Barr virus; HLA: human leukocyte antigen; EA: early antigen; LP: leading protein; EBER: small RNA encoded by EBV; IL: interleukin; BHRF: functional counterpart of Bcl-2; BART: EBV microRNA; IRES: internal ribosome entry site; BMRF: viral cell adhesion protein; GP: glycoprotein; IFN-γ: interferon gamma; LMP: latent membrane protein; NF-κβ: nuclear factor kappa-light-chain-enhancer of activated B cells.
Source: Own elaboration based on Tracy et al. (3), Kang & Kieff (15), Maghzi et al. (17), Moss et al. (19), Isaksson et al. (20), Shinozaki-Ushiku et al. (21) and Iwakiri & Takada (22).
During latency stages, EBV expresses genes only to survive and prolong the infection. Cells with latent infection express low immunogenicity proteins according to the latency stage expressed by EBV (Table 3). Viral infection persists throughout life and progresses with intermittent lytic reactivation. (4,23-25)
In primary infection, the virus infects immune cells and epithelial cells while activating its type III latency genes complex. The infected B cells then enter the lymph nodes, where they proliferate and express viral proteins corresponding to type II latency. Thanks to the type 0 latency proteins, infected B cell reach circulation and undergo negative regulation by EBV to evade the immune response. Then, they divide and express a single viral protein (EBNA-1, type I latency) that ensures the division of the viral genome. Once these cells pass through the oropharynx, they transfer the virus to the epithelial cells, where it replicates to infect other cells (Figure 2). (26,27)
Table 3. Stages of Epstein-Barr virus infection.
|
Type of infection |
Description |
|
Latency type III (growth) |
Initial phase of infection in B cells, where EBV activates growth programs. Expression of all EBNA and LMP. |
|
Latency type II (default) |
Expression of EBNA-1, LMP-1 and LMP-2. It allows the transformation of blasts into memory B cells in the tonsillar germinal centers. Once differentiated, they can reside there or go to peripheral circulation. Inactivation of the other EBV genes. |
|
Latency type I |
Once in circulation, infected B cells do not express proteins except during cell division (S phase), when they only activate EBNA-1. Thus, they facilitate the transfer of the viral genome to the daughter cells. B cells with latent infection type I are transformed into plasma cells. |
|
Lytic infection cycle |
EBV replicates its genome, elaborates structural components, and lyses B cells and epithelial cells that contain viral copies. |
EBV: Epstein-Barr virus; EBNA: EBV nuclear antigen; LMP: latent membrane protein.
Source: Own elaboration based on Kang & Kief (15) and Füst (16).

Figure 2. Epstein-Barr virus latency stages in the host.
Source: Own elaboration.
EBV and autoimmune diseases
During the normal maturation process, immune cells learn to differentiate their own antigens (autoantigens) from anything that is foreign (antigens). The axis of immunological tolerance is the TCreg (CTCD4+), whose main function is to maintain tissue homeostasis through the suppression of inflammatory responses using four mechanisms: 1) regulation of antigen presentation, 2) destruction of target cells, 3) alteration of metabolic pathways, and 4) production of anti-inflammatory cytokines. (28,29) When these mechanisms are altered, hypersensitivity reactions and consequent autoimmune diseases are more likely to occur in predisposed individuals. (3) Therefore, tolerance to autoantigens is lost and the immune system no longer recognizes its own cells or tissues, which results in the inmune system attacking its own cells. The immediate cause of autoimmune diseases is not clear; however, there is evidence of a complex interaction between genetic, epigenetic and environmental risk factors that favor their presentation. (30,31)
Among the environmental factors associated with the pathogenesis of autoimmune diseases, infections top the list, and EBV, in particular, has been associated with the development of these diseases. (4) It is well known that infectious agents can initiate inflammatory responses by adhering to Toll-like receptors (TLRs), antibodies, complement cascade fractions, mannose-dependent lectins, or intracellular receptors. (32) EBV induces the production of pro-inflammatory cytokines and autoantibodies that stimulate hyperactive B cells and T cells to abandon their anergic states. In this way, the immune system can initiate and perpetuate the inflammatory response against autoantigens by cross-reacting with viral antigens (molecular mimicry) and residues of cellular and viral metabolism. (33-35) Flow cytometry and real-time PCR have demonstrated that viral RNA transcripts (EBER-1 and -2) in cells with latent infection adhere to protein kinase RNA-activated (PKR), ribosomal protein 22 (L22), antigen associated with SLE (La), and retinoic acid inducible gene I (RIG-I). (36) This promotes the production of type I IFNs, pro-inflammatory cytokines and autoantibodies via the TLR3 signaling pathway. (19,34,36)
There is a large number of studies that have addressed the relationship between EBV and the development of autoimmune diseases such as systemic lupus erythematosus (37,38) and its exacerbations (39); rheumatoid arthritis (40); type I diabetes mellitus (41); Sjogren’s syndrome (42); autoimmune thyroiditis (43); autoimmune hepatitis (44); systemic sclerosis (18); transplant rejection due to viral reactivation (45); autoimmune heart disease (46-48); adverse drug reactions (49,50); Parkinson’s disease (51); and MS. (3,10)
Multiple sclerosis
MS is an autoimmune disease of the central nervous system with a course that is difficult to predict in each patient. It is characterized by multiple foci of inflammation, demyelination, axonal injury, fibrosis, grey matter involvement and neurodegeneration. (52,53) Nearly half of MS patients suffer from anxiety disorders, depression, bipolarity and cognitive dysfunction at some point in their lives. Several studies have shown that cerebral atrophy is associated with MS, leading to a cognitive and motor deficit evidenced by a reduction in the volume of the prefrontal, parietal and temporal cortex. (7,54)
MS is the leading cause of disability among young adults. About 90% of those affected have the relapsing-remitting form of the disease, 10% present with a primary progressive form, while the secondary progressive form is observed to a lesser extent. (55) The peak age of onset is 30 years, although it can be diagnosed in children and adults over 60. (55) MS occurs most frequently in people aged 25-54 and represents an important economic burden for patients, their families, the health system and society in general. (56)
Epidemiology of MS
MS affects more than 2.5 million people worldwide (57); while the primary progressive form affects men and women equally, females are more susceptible (3:1) to the relapsing-remitting form. (58) There is a relatively high prevalence and incidence of MS in North America and Europe, areas with the the largest Caucasian population proportions, the highest income rates and better access to health care. For example, in the British Isles, its prevalence is 96 per 100 000 inhabitants, while its incidence is 7.2-12.2 per 100 000 inhabitants, while in Italy, the prevalence is 100 per 100 000 inhabitants and the incidence, 3.4-6.8 per 100 000 inhabitants. (59) Likewise in USA, MS prevalence ranges from 39.9 to 191.2 cases per 100 000 inhabitants and the MS incidence is 7.3 per 100 000 inhabitants. (59) On the other hand, the lowest MS prevalence and incidence rates are found in mestizo populations living in tropical and subtropical regions: in Latin America, MS prevalence ranges from 1.6 to 19.6 per 100 000 inhabitants and the incidence is 1.4 per 100 000 inhabitants (60.61), while in Colombia, MS prevalence is 3 per 100 000 inhabitants. (61)
There is a relatively high prevalence and incidence of MS in Europe and North America, areas with the largest Caucasian population, highest income and improved access to health care. On the other hand, the mestizo populations (white and Amerindian) living in tropical and subtropical regions have the lowest prevalence and incidence rates of this disease. (61)
Genetic risk factors for MS and EBV-MS association
EBV-positive individuals with alterations in HLA alleles have a seven-fold increased risk of developing MS compared with carriers who do not. (10) The HLA-DRB1 and HLA-DRQ1 genes are strongly associated with susceptibility and progression of MS. There are also genes outside the HLA region directly related to cellular immunity functions that may be associated with the pathogenesis of MS. (59)
Self-reactive memory B cells and T cells infected by EBV are activated at the periphery and cross the blood-brain barrier (BBB) by increased vascular permeability and underlying an inflammatory response (Th1/Th2/Th17). In the neuroparenchyma, T cells secrete cytokines (IL-2, IL-17, IFNγ, TNFα), elevating their concentration in the cerebrospinal fluid; they also present the myelin antigen (MA) to the microglia arranged around the myelin sheaths of the oligodendrocytes and attacks them. Circulating memory B cells migrate to the neuroparenchyma and differentiate into plasma cells (PC) that produce autoantibodies against MA (MBP: myelin basic protein, MOG: myelin oligodendrocytic glycoprotein, MAG: myelin-associated glycoprotein, PLP: proteolipid protein) that aggravate and prolong neuroinflammation. After the outbreak of MS, repair mechanisms are activated with subsequent fibrosis (Figure 3). (4,5,52,62)
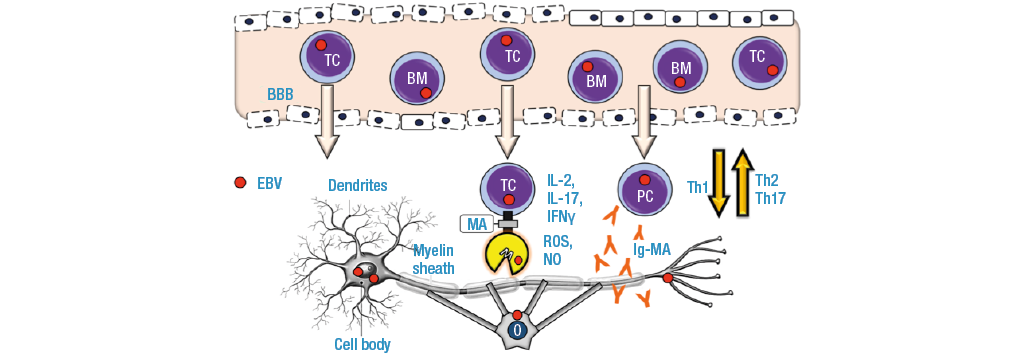
Figure 3. Patogénesis de la esclerosis múltiple.
BBB: blood brain barrier; TC: T cell; BM: B Memory cell; PC: plasma cell; M: microglia; MA: myelin antigen; O: oligodendrocite; Ig-MA: myelin antigen antibody; IL: interleukin; IFN: interferon; ROS: reactive oxygen species; NO: nitric oxide.
Source: Own elaboration.
Zhou et al. (11) determined genetic loci that influence anti-EBNA-1 IgG titers and may play an important role in the predisposition to develop MS. These researchers conducted a genome-wide association study (GWAS) to assess the polygenic risk shared between EBNA-1 and MS in 5 555 and 15 231 individuals, respectively. They identified a locus that showed a strong association in the HLA gene. The most significantly associated genotype was single nucleotide polymorphism (SNP) rs25160.49 (p=4.11 x 10-9). This finding is consistent with a GWAS performed on a cohort of Mexican-American families, in which the same SNP rs2516049 was confirmed (p=3.32 x 10-20). The authors concluded that the association of the HLA region and non-HLA genes (antiEBNA-1 IgG) are positively related to the development of MS. (11)
It is considered that HLA alleles may favor molecular mimicry between EBV proteins, especially EBNA-1 (52), and autoantigens such as MBP through the recognition and presentation of T cells. (63-65) Hyperreactivity against MOG (9,66), MAG (62,67) and PLP (68,69) have also been studied in several demyelinating diseases, and their association with MS has been the most studied.
EBNA-1 favors cross-reaction with myelin antigens, so there is an increase in IFN-γ and IL-2. Anti-EBNA-1 antibodies increase in the presence of clinical manifestations of MS and during disease progression. (52,70-72) Humoral immunity plays an important role in MS, revealing IgG production against intrathecal epitopes of EBV, while some patients present highly reactive oligoclonal bands against viral protein in the cerebrospinal fluid study. (70,73)
The evidence involving EBV in the pathogenesis of MS has increased over the years: 99.5% of MS patients are seropositive for EBV, although control populations have a high rate of asymptomatic infection as well (94.2%). (8,74) A history of infectious mononucleosis is associated with the presentation of autoimmune diseases and an increased risk of MS (75), since there is massive viral replication in 50% of young adult patients that favors persistent stimulation of the immune system with chronic production of autoantibodies that, in turn, increase the likelihood of hypersensitivity reactions against autoantigens. It is interesting that antibodies against EBV proteins (BRRF2 and EBNA-1) are observed, being significantly higher in serum and CSF of patients with MS than in controls. Similarly, the cytotoxic response to latent viral proteins is greater in patients than in controls. (76,77)
Patients with a history of infectious mononucleosis are 20 times more likely to progress to MS than EBV-negative patients. (78) Likewise, Thacker et al. (79) suggest that those patients who previously presented with infectious mononucleosis have twice the risk of developing MS [relative risk (RR) 2.3, 95%CI: 1.7-3.0, p<10-8]. (79) Handel et al. (80), in a meta-analysis, calculated an approximate RR of 2.17 (95%CI: 1.97-2.39, p<10-54). In patients with MS, most B cells and plasma cells are infected with EBV, and 99% are seropositive for EBV; therefore, it has been proposed that vaccination could be a disease prevention measure. (57)
Conclusions
Depending on the condition of the immune system and the predisposing genetic traits of each individual, there may be interactions with EBV that predispose it to trigger intense inflammatory responses and, by molecular mimicry, affect cells and nerve tissues. Taking this into account, intense inflammatory responses can cause various clinical manifestations of MS.
More studies addressing the pathogenesis,the morphophysiological changes and the neuroimmunological alterations produced by the EVB-inmmune response coplex at the molecular, cellular, tissue, organic and systemic level are needed.
Seroprevalence studies in Colombia are suggested in order to know the epidemiology of EBV and other herpesviruses. With this in mind, research involving professionals of the basic and clinical sciences, epidemiology and public health can be guided. Studies with larger populations, of longer duration and analyzing risk factors and clinical variables, in addition to those regularly described among MS patients, are also required. This could have potential applications in clinical management and promotion-prevention measures, diagnosis, prognosis and research in the development of therapeutic resources.
Conflicts of interest
None stated by the authors.
Funding
None stated by the authors.
Acknowledgements
None stated by the authors.
References
1.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964;1(7335):702-3. http://doi.org/dmszkn.
2.Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt’s lymphoma: a polymicrobial disease? Nat Rev Microbiol. 2005;3(2):182-7. http://doi.org/cqmnb7.
3.Tracy SI, Kakalacheva K, Lünmeann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. Persistence of Epstein-Barr Virus in Self-Reactive Memory B Cells. J Virol. 2012;86(22):12330-40. http://doi.org/f4bf7t.
4.Draborg AH, Duus K, Houen G. Epstein-Barr Virus in Systemic Autoimmune Diseases. Clin Dev Immunol. 2013;2013:535738. http://doi.org/f9snfz.
5.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23(1):683-747. http://doi.org/d8tp56.
6.Cencioni MT, Magliozzi R, Nicholas R, Ali R, Malik O, Reynolds R, et al. Programmed death 1 is highly expressed on CD8+ CD57+ T cells in patients with stable multiple sclerosis and inhibits their cytotoxic response to Epstein-Barr virus. Immunology. 2017;152(4):660-76. http://doi.org/gbx6jx.
7.Kapadia M, Sakic B. Autoimmune and inflammatory mechanisms of CNS damage. Prog Neurobiol. 2011;95(3):301-33. http://doi.org/bvdzm6.
8.Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernán MA, Olek MJ, et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: A prospective study. JAMA. 2001;286(24):3083-8. http://doi.org/d58stf.
9.Kakalacheva K, Regenass S, Wiesmayr S, Azzi T, Berger C, Dale RC, et al. Infectious Mononucleosis Triggers Generation of IgG Auto-Antibodies against Native Myelin Oligodendrocyte Glycoprotein. Viruses. 2016;8(2):E51. http://doi.org/f8pnvr.
10.Casiraghi C, Dorovini-Zis K, Horwitz MS. Epstein-Barr virus infection of human brain microvessel endothelial cells: A novel role in multiple sclerosis. J Neuroimmunol. 2011;230(1-2):173-7. http://doi.org/fk9bb6.
11.Zhou Y, Zhu G, Charlesworth JC, Simpson S, Rubicz R, Göring HH, et al. Genetic loci for Epstein-Barr virus nuclear antigen-1 are associated with risk of multiple sclerosis. Mult Scler. 2016;22(13):1655-64. http://doi.org/f9h69q.
12.Dowd JB, Palermo T, Brite J, McDade TW, Aiello A. Seroprevalence of Epstein-Barr Virus Infection in U.S. Children Ages 6-19, 2003-2010. PLoS One. 2013;8(5):e64921. http://doi.org/c2rm.
13.Odumade OA, Hogquist KA, Balfour HH. Progress and problems in understanding and managing primary epstein-barr virus infections. Clin Microbiol Rev. 2011;24(1):193-209. http://doi.org/bgdhwn.
14.International Committee on Taxonomy of Viruses (ICTV). ICTV Taxonomy history: Human gammaherpesvirus 4. 2016 [cited 2019 Abr 17]. Available from: https://goo.gl/5Jh2ay.
15.Kang MS, Kieff E. Epstein-Barr virus latent genes. Exp Mol Med. 2015;47(1):e131. http://doi.org/c2rn.
16.Füst G. The role of the Epstein-Barr virus in the pathogenesis of some autoimmune disorders - Similarities and differences. Eur J Microbiol Immunol. 2011;1(4):267-78. http://doi.org/fzhrxz.
17.Maghzi AH, Marta M, Bosca I, Etemadifar M, Dobson R, Maggiore C, et al. Viral pathophysiology of multiple sclerosis: A role for Epstein-Barr virus infection? Pathophysiology. 2011;18(1):13-20. http://doi.org/dwfkcs.
18.Farina A, Peruzzi G, Lacconi V, Lenna S, Quarta S, Rosato E, et al. Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res Ther. 2017;19(1):39. http://doi.org/c2rp.
19.Moss WN, Lee N, Pimienta G, Steitz JA. RNA families in Epstein-Barr virus. RNA Biol. 2014;11(1):10-7. http://doi.org/c2rq.
20.Isaksson A, Berggren M, Ricksten A. Epstein-Barr virus U leader exon contains an internal ribosome entry site. Oncogene. 2003;22(4):572-81. http://doi.org/c7jv2s.
21.Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein-Barr virus and gastric cancer (Review). Int J Oncol. 2015;46(4):1421-34. http://doi.org/c2rr.
22.Iwakiri D, Takada K. Role of EBERs in the Pathogenesis of EBV Infection. Adv Cancer Res. 2010;107:119-36. http://doi.org/fdn9sq.
23.Lossius A, Johansen JN, Torkildsen Ø, Vartdal F, Holmøy T. Epstein-Barr Virus in Systemic Lupus Erythematosus, Rheumatoid Arthritis and Multiple Sclerosis—Association and Causation. Viruses. 2012;4(12):3701-30. http://doi.org/gcfn25.
24.Libbey JE, Cusick MF, Fujinami RS. Role of Pathogens in Multiple Sclerosis. Int Rev Immunol. 2014;33(4):266-83. http://doi.org/c2rs.
25.Owens GP, Bennett JL. Trigger, pathogen, or bystander: the complex nexus linking Epstein-Barr virus and multiple sclerosis. Mult Scler. 2012;18(9):1204-8. http://doi.org/f37z73.
26.Medina-Ortega ÁP, López-Valencia D, Mosquera-Monje SL, Mora-Obando DL, Dueñas-Cuéllar RA. Virus de Epstein-Barr y su relación con el desarrollo del cáncer. Iatreia. 2017;30(2):131-45. http://doi.org/c2rt.
27.Bollard CM, Rooney CM, Heslop HE. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol. 2012;9(9):510-9. http://doi.org/c2rv.
28.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523-32. http://doi.org/cnjn86.
29.Siachoque H, Satisteban N, Iglesias-Gamarra A. Linfocitos T reguladores: Subpoblaciones, mecanismo de acción e importancia en el control de la autoinmunidad. Rev Colomb Reumatol. 2011;18(3):203-20. http://doi.org/f2mdmp.
30.Zhao M, Wang Z, Yung S, Lu Q. Epigenetic dynamics in immunity and autoimmunity. Int J Biochem Cell Biol. 2015;67:65-74. http://doi.org/c2rw.
31.Doria A, Sarzi-Puttini P, Shoenfeld Y. Infections, rheumatism and autoimmunity: The conflicting relationship between humans and their environment. Autoimmun Rev. 2008;8(1):1-4. http://doi.org/cmsxzt.
32.Kumar V, Abbas A, Fausto N, Aster J. Inflamación aguda y crónica. In: Patología estructural y funcional. 8th ed. Barcelona: Elsevier; 2010. p. 43-77.
33.Williams MV, Cox B, Ariza ME. Herpesviruses dUTPases: A New Family of Pathogen-Associated Molecular Pattern (PAMP) Proteins with Implications for Human Disease. Pathogens. 2016;6(1):2. http://doi.org/c2rx.
34.Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, et al. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from toll-like receptor 3. J Exp Med. 2009;206(10):2091-9. http://doi.org/cwmqcr.
35.Pawaria S, Sharma S, Baum R, Nündel K, Busto P, Gravallese ME, et al. Taking the STING out of TLR-driven autoimmune diseases: good, bad, or indifferent? J Leukoc Biol. 2017;101(1):121-6. http://doi.org/f9nckb.
36.Fok V, Mitton-Fry RM, Grech A, Steitz JA. Multiple domains of EBER 1, an Epstein-Barr virus noncoding RNA, recruit human ribosomal protein L22. RNA. 2006;12(5):872-82. http://doi.org/ft7gmv.
37.Abdel-Wahab N, Talathi S, López-Olivo MA, Suárez-Almazor ME. Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus. 2018;27(4):572-83. http://doi.org/c2rz.
38.Chougule D, Nadkar M, Rajadhyaksha A, Pandit-Shende P, Surve P, Dawkar N, et al. Association of clinical and serological parameters of systemic lupus erythematosus patients with Epstein-Barr virus antibody profile. J Med Virol. 2018;90(3):559-63. http://doi.org/c2r2.
39.Draborg AH, Lydolph MC, Westergaard M, Olesen Larsen S, Nielsen CT, Duus K, et al. Elevated Concentrations of Serum Immunoglobulin Free Light Chains in Systemic Lupus Erythematosus Patients in Relation to Disease Activity, Inflammatory Status, B Cell Activity and Epstein-Barr Virus Antibodies. PLoS One. 2015;10(9):e0138753. http://doi.org/c2r3.
40.Mehraein Y, Lennerz C, Ehlhardt S, Remberger K, Ojak A, Zang KD. Latent Epstein-Barr virus (EBV) infection and cytomegalovirus (CMV) infection in synovial tissue of autoimmune chronic arthritis determined by RNA- and DNA-in situ hybridization. Mod Pathol. 2004;17(7):781-9. http://doi.org/bgf8bz.
41.Faustman DL. EBV infection and anti-CD3 treatment for Type 1 diabetes: bad cop, good cop? Expert Rev Clin Immunol. 2013;9(2):95-7. http://doi.org/c2r4.
42.Croia C, Astorri E, Murray-Brown W, Willis A, Brokstad KA, Sutcliffe N, et al. Implication of Epstein-Barr Virus Infection in Disease-Specific Autoreactive B Cell Activation in Ectopic Lymphoid Structures of Sjögren’s Syndrome. Arthritis Rheumatol. 2014;66(9):2545-57. http://doi.org/f6gkn3.
43.Nagata K, Nakayama Y, Higaki K, Ochi M, Kanai K, Matsushita M, et al. Reactivation of persistent Epstein-Barr virus (EBV) causes secretion of thyrotropin receptor antibodies (TRAbs) in EBV-infected B lymphocytes with TRAbs on their surface. Autoimmunity. 2015;48(5):328-35. http://doi.org/c2r5.
44.Yamashita H, Shimizu A, Tsuchiya H, Takahashi Y, Kaneko H, Kano T, et al. Chronic active Epstein-Barr virus infection mimicking autoimmune hepatitis exacerbation in a patient with systemic lupus erythematosus. Lupus. 2014;23(8):833-6. http://doi.org/f59qb9.
45.Mesa JG, Aristizábal BH. Seguimiento con carga viral para virus Epstein-Barr en pacientes pediátricos con trasplante hepático. Médicas UIS. 2015;28(3):393-401. http://doi.org/c2r6.
46.Pankuweit S, Klingel K. Viral myocarditis: from experimental models to molecular diagnosis in patients. Heart Fail Rev. 2013;18(6):683-702. http://doi.org/f5hjkd.
47.Roubille F, Gahide G, Moore-Morris T, Granier M, Davy JM, Vernhet H, et al. Epstein Barr Virus (EBV) and Acute Myopericarditis in an Immunocompetent Patient: First Demonstrated Case and Discussion. Intern Med. 2008;47(7):627-9. http://doi.org/bh76vf.
48.Kawamura Y, Miura H, Matsumoto Y, Uchida H, Kudo K, Hata T, et al. A case of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis with severe cardiac complications. BMC Pediatr. 2016;16(1):172. http://doi.org/f9bk66.
49.Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A. 1995;92(16):7440-4. http://doi.org/cb55z2.
50.Shiohara T, Inaoka M, Kano Y. Drug-induced Hypersensitivity Syndrome (DIHS): A Reaction Induced by a Complex Interplay among Herpesviruses and Antiviral and Antidrug Immune Responses. Allergol Int. 2006;55(1):1-8. http://doi.org/bwrzt7.
51.Woulfe JM, Gray MT, Gray DA, Munoz DG, Middeldorp JM. Hypothesis: A role for EBV-induced molecular mimicry in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(7):685-94. http://doi.org/f59hvn.
52.Pender MP, Burrows SR. Epstein-Barr virus and multiple sclerosis: potential opportunities for immunotherapy. Clin Transl Immunol. 2014;3(10):e27. http://doi.org/c2r7.
53.Kakalacheva K, Lünmeann JD. Environmental triggers of multiple sclerosis. FEBS Lett. 2011;585(23):3724-9. http://doi.org/c6v7p7.
54.Racke MK, Imitola J. Cortical Volume Loss and Neurologic Dysfunction in Multiple Sclerosis. JAMA Neurol. 2016;73(8):910-2. http://doi.org/c2r8.
55.Raffel J, Wakerley B, Nicholas R. Multiple sclerosis. Medicine. 2016;44(9):537-41. http://doi.org/c2r9.
56.Chung SE, Cheong HK, Park JH, Kim HJ. Burden of Disease of Multiple Sclerosis in Korea. Epidemiol Health. 2012;34:e2012008. http://doi.org/c2sb.
57.Pender MP. The Essential Role of Epstein-Barr Virus in the Pathogenesis of Multiple Sclerosis. Neuroscientist. 2011;17(4):351-67. http://doi.org/fwxnp9.
58.Sellner J, Kraus J, Awad A, Milo R, Hermer B, Stüve O. The increasing incidence and prevalence of female multiple sclerosis—A critical analysis of potential environmental factors. Autoimmun Rev. 2011;10(8):495-502. http://doi.org/cpz654.
59.Howard J, Trevick S, Younger DS. Epidemiology of Multiple Sclerosis. Neurol Clin. 2016;34(4):919-39. http://doi.org/c2sc.
60.Melcon M, Gold L, Carrá A, Cáceres F, Correale J, Cristiano E, et al. Argentine Patagonia: prevalence and clinical features of multiple sclerosis. Mult Scler J. 2008;14(5):656-62. http://doi.org/fgfw2j.
61.Ojeda E, Díaz-Cortes D, Rosales D, Duarte-Rey C, Anaya JM, Rojas-Villarraga A. Prevalence and clinical features of multiple sclerosis in Latin America. Clin Neurol Neurosurg. 2013;115(4):381-7. http://doi.org/f4svnk.
62.Langkamp M, Hörnig SC, Hörnig JB, Kirschner M, Pridzun L, Kornhuber ME. Detection of myelin autoantibodies: evaluation of an assay system for diagnosis of multiple sclerosis in differentiation from other central nervous system diseases. Clin ChME Lab Med. 2009;47(11):1395-400. http://doi.org/bn6b3c.
63.Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. 2002;3(10):940-3. http://doi.org/c7j7mk.
64.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80(5):695-705. http://doi.org/dqf4jw.
65.Friedrich MG, Hancock SE, Raftery MJ, Truscott RJ. Isoaspartic acid is present at specific sites in myelin basic protein from multiple sclerosis patients: could this represent a trigger for disease onset? Acta Neuropathol Commun. 2016;4(1):83. http://doi.org/c2tr.
66.Thulasirajah S, Pohl D, Davila-Acosta J, Venkateswaran S. Myelin Oligodendrocyte Glycoprotein-Associated Pediatric Central Nervous System Demyelination: Clinical Course, Neuroimaging Findings, and Response to Therapy. Neuropediatrics. 2016;47(4):245-52. http://doi.org/f9p9x4.
67.Kuhlmann T, Ludwin S, Prat A, Antel J, Brück W, Lassmann H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017;133(1):13-24. http://doi.org/f9m72m.
68.Rühl G, Niedl AG, Patronov A, Siewert K, Pinkert S, Kalemanov M, et al. Multiple sclerosis: Molecular mimicry of an antimyelin HLA class I restricted T-cell receptor. Neurol Neuroimmunol Neuroinflamm. 2016;3(4):e241. http://doi.org/c2ts.
69.Harlow DE, Saul KE, Komuro H, Macklin WB. Myelin Proteolipid Protein Complexes with αv Integrin and AMPA Receptors In Vivo and Regulates AMPA-Dependent Oligodendrocyte Progenitor Cell Migration through the Modulation of Cell-Surface GluR2 Expression. J Neurosci. 2015;35(34):12018-32. http://doi.org/c2tt.
70.Pender MP. Preventing and curing multiple sclerosis by controlling Epstein-Barr virus infection. Autoimmun Rev. 2009;8(7):563-8.
http://doi.org/bjw24z.
71.Deeba E, Koptides D, Gaglia E, Constantinou A, Lambrianides A, Pantzaris M, et al. Evaluation of Epstein-Barr virus-specific antibodies in Cypriot multiple sclerosis patients. Mol Immunol. 2019;105:270-5. http://doi.org/c4m3.
72.Jakimovski D, Ramanathan M, Weinstock-Guttman B, Bergsland N, Ramasamay DP, Carl E, et al. Higher EBV response is associated with more severe gray matter and lesion pathology in relapsing multiple sclerosis patients: A case-controlled magnetization transfer ratio study. Mult Scler. 2019. http://doi.org/c4m4.
73.Lindsey JW, Hatfield LM. Epstein-Barr virus and multiple sclerosis: Cellular immune response and cross-reactivity. J Neuroimmunol. 2010;229(1-2):238-42. http://doi.org/b36xsw.
74.Goodin DS. The Causal Cascade to Multiple Sclerosis: A Model for MS Pathogenesis. PLoS One. 2009;4(2):e4565. http://doi.org/bcm643.
75.Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. 2010;6(12):683-92. http://doi.org/b8j7ps.
76.Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Büssow K, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115(5):1352-60. http://doi.org/b95h73.
77.Lossius A, Johansen JN, Vartdal F, Robins H, Jūratė Šaltytė B, Holmøy T, et al. High-throughput sequencing of TCR repertoires in multiple sclerosis reveals intrathecal enrichment of EBV-reactive CD8+ T cells. Eur J Immunol. 2014;44(11):3439-52. http://doi.org/f2t892.
78.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann Neurol. 2007;61(4):288-99. http://doi.org/d8dq3d.
79.Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: A meta-analysis. Ann Neurol. 2006;59(3):499-503. http://doi.org/ffj4pp.
80.Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One. 2010;5(9):e12496. http://doi.org/c6ddp8.

Jean-Baptiste Marc Bourgery. (1797- 1849)
“Traité complet de l’anatomie de l’homme: comprenant la médecine operatoire”