case report
DOI: https://doi.org/10.15446/revfacmed.v67n3.70507
Late recurrence of adrenocortical carcinoma and metastatic disease. Case report
Recurrencia tardía de carcinoma adrenocortical con compromiso metastásico. Reporte de un caso
Received: 17/02/2018. Accepted: 23/03/2018.
María Alejandra Pérez-Ardila1 • Julián Naranjo-Millán1 • Helman Giral1 • Jaqueline Mugnier2 • Henry Altamar3
1 Fundación Cardioinfantil - Cardiology Institute - Department of Internal Medicine - Bogotá D.C. - Colombia.
2 Fundación Cardioinfantil - Cardiology Institute - Department of Pathology - Bogotá D.C. - Colombia.
3 Fundación Cardioinfantil - Cardiology Institute - Department of Endocrinology - Bogotá D.C. - Colombia.
Corresponding author: María Alejandra Pérez-Ardila. Departamento de Medicina Interna, Fundación Cardioinfantil - Instituto de Cardiología. Calle 163A No 13B-60, Office of Internal Medicine - Staff Tower. Phone number: +57 1 6672727, ext.: 55304; Mobile number: +57 3164224089. Bogotá D.C. Colombia. Email: mariaperezardila@gmail.com.
| Abstract |
Introduction: Adrenocortical carcinoma (ACC) is a malignancy with an annual incidence of 0.72 cases per million people. It is a rare tumor that is associated with high mortality in late stages, as well as with a 5-year survival of 13% in stage IV patients, and 61% in stage II lesions. Nevertheless, tumor recurrence occurs in up to 54% at 23 months with predominance of locoregional involvement, being striking the late presentation of tumor recurrence with extensive involvement.
Case presentation: This is the case of a 52-year-old male patient with a history of resection of an ACC of 6cm five years earlier, who was admitted for decompensated heart failure. A chest x-ray was taken and metastasis was suspected. After conducting biochemical studies and a CT scan of the abdomen, ACC with metastases to the liver and lung was diagnosed. The patient decided not to receive palliative chemotherapy.
Conclusion: This unfortunate outcome is related to the lack of follow-up after the initial procedure. Clinical, hormonal, and imaging evaluation is recommended every 3 months for 2 years, and then at least every 6 months.
Keywords: Adrenocortical Carcinoma; Adrenal Cortex Neoplasms; Adrenal Gland Neoplasms (MeSH).
Pérez-Ardila MA, Naranjo-Millán J, Giral H, Mugnier J, Altamar H. Late recurrence of adrenocortical carcinoma and metastatic disease. Case report. Rev. Fac. Med. 2019;67(3):525-9. English. doi https://doi.org/10.15446/revfacmed.v67n3.70507.
| Resumen |
Introducción. El carcinoma adrenocortical (CAC) es una neoplasia que reporta incidencia anual de 0.72 casos por cada millón de personas. Se trata de un tumor infrecuente que se asocia con una mortalidad elevada en estadios avanzados y una supervivencia a 5 años del 13% de pacientes en estadio IV y del 61% para lesiones en estadio II; sin embargo, la presencia de recurrencia tumoral es hasta de 54% a los 23 meses con predominio de compromiso locoregional, siendo llamativa la presentación tardía de la recurrencia tumoral con compromiso extenso.
Presentación del caso. Paciente masculino de 52 años con antecedente de resección de un CAC de 6cm de tamaño 5 años atrás, quien ingresa por falla cardíaca descompensada, encontrando en la radiografía de tórax una imagen sugestiva de metástasis. Tras estudios bioquímicos y tomografía de abdomen se diagnostica CAC con compromiso metastásico a hígado y pulmón. El paciente opta por no recibir manejo quimioterapéutico paliativo.
Conclusión. Este desenlace desafortunado tiene relación con la ausencia de seguimiento tras el procedimiento inicial. Se recomienda evaluación clínica, hormonal e imagenológica cada 3 meses por 2 años y luego al menos cada 6 meses.
Palabras clave: Neoplasias de la corteza suprarrenal; Enfermedades de la corteza suprarrenal; Glándulas suprarrenales (DeCS).
Pérez-Ardila MA, Naranjo-Millán J, Giral H, Mugnier J, Altamar H. [Recurrencia tardía de carcinoma adrenocortical con compromiso metastásico. Reporte de un caso]. Rev Fac. Med. 2019;67(3):525-9. English. doi: https://doi.org/10.15446/revfacmed.v67n3.70507.
Introduction
Adrenal tumors affect 3-10% of the world’s population; specifically, adrenocortical carcinoma (ACC) has a prevalence of <200 000 in the U.S., with an estimated annual incidence of 0.72 cases per million people, with a higher number of cases in the female sex. (1,2)
ACC has a bimodal distribution and is more common in the first and fifth decades of life. Half of the patients are asymptomatic or have mechanical symptoms caused by the size of the tumor, while the other half may have symptoms due to hormonal production (3); this hormonal release may result in hypercortisolism, hyperaldosteronism, hyperandrogenism or production of several hormones simultaneously. (4)
The objective of this article is to present a case that highlights the importance of evaluating malignancy characteristics in adrenal masses, and the consequences of inadequate surgical management. Besides reporting this case, a brief review on ACC is also presented.
Case presentation
This is the case of a 52-year old male patient who attended consultation due to an edema of 20 days of evolution in the lower limbs, associated with a decrease in functional class, which used to be NYHA I and changed to NYHA II at the time of admission.
Upon completion of the medical history, the patient also reported 12kg weight gain in 3 months, as well as decreased libido and difficulty ejaculating. The man had a history of a tumor in the right adrenal gland, for which right adrenalectomy by laparoscopy had been performed 5 years earlier (year 2012). The surgical report was not available, but the patient reported that the tumor was 8cm in diameter. The pathology described a tumor of cortical origin, with 20% Ki-67 and 3 Weiss criteria that indicated malignancy.
The laboratory tests in his previous medical chart showed elevation of FSH, LH and total testosterone (Table 1). The patient did not receive adjuvant management for the adrenal carcinoma. A diagnosis of adrenal insufficiency requiring mineralocorticoid replacement was also made after the procedure.
Table 1. Previous hormonal profile of the patient
|
Date Test |
June 2013 |
January 2014 |
April 2014 |
August 2014 |
|
Aldosterone (10-160 pg/mL) |
35.7 pg/mL |
|||
|
Total renin (1.68-27.66 pg/mL) |
100.60 pg/mL |
|||
|
FSH (0.95-11.9 mUI/mL) |
13.9 mUI/mL |
12.13 mUI/mL |
||
|
LH (0.57-12.07mUI/mL) |
19.6mUI/mL |
16.94 mUI/mL |
||
|
Total testosterone (1.42-9.23ng/mL) |
4.07ng/mL |
14.76 ng/mL |
||
|
DHEA (136.2-447.6ug/dL) |
13.6 ug/dL |
23.2 ug/dL |
||
|
Cortisol in urine |
0.00 |
0.00 |
0.00 |
0.00 |
|
Vanillylmandelic acid (1-13.6mg/24 hours) |
6 mg/24 hours |
8.6 mg/24 hours |
||
|
Calcitonin |
<2 pg/mL |
|||
|
PTH (11-67 pg/mL) |
22 pg/mL |
FSH: follicle stimulating hormone; LH: luteinizing hormone; PTH: parathyroid hormone; DHEA: dehydroepiandrosterone.
Source: Own elaboration.
On admission, the patient presented tachycardia, blood pressure of 186/117 mmHg, body mass index of 26.7 kg/m2, cushingoid facies, central obesity, gynecomastia, ecchymosis in the arms (without skin atrophy), signs of congestion such as jugular vein distention and bilateral edema in the lower limbs. No alterations in hemogram, renal function, electrolytes or liver function were observed in the admission tests.
Taking into account the dyspnea referred by the patient, a chest x-ray was performed which showed cardiomegaly and a radiopaque lesion of 37x38mm in the right upper lobe, with well-defined edges. In addition, a transthoracic echocardiogram showed hypokinesia of the left ventricle with ejection fraction of 33% and signs of moderate aortic insufficiency.
Initially, management was given to compensate for heart failure. Considering his medical history and the presence of a mass in the upper right lobe, a computed tomography (CT) of the thorax was performed in which a mass in the upper right lobe of 38x29x29mm was observed, without enhancement after the administration of contrast medium. It also showed three other nodular images with the same characteristics in the apical segment of the upper left lobe with a secondary neoplastic aspect (Figure 1).
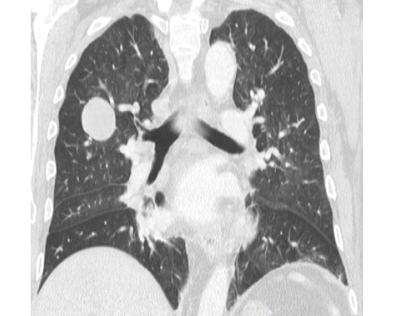
Figure 1. CT scan of the chest.
Source: Document obtained during the study.
A magnetic resonance (MRI) of the abdomen showed a large retroperitoneal mass of 19x18x13cm, which displaced the ascending colon, the second duodenal portion, and the head of the pancreas. The lesion had a heterogeneous signal pattern in sequences with T1 and T2 information, predominantly hyperintense, with heterogeneous enhancement after administration of contrast medium. These findings suggested solid lesion with associated hemorrhagic residuals and multiple focal liver lesions that had a similar pattern (Figure 2).

Figure 2. Contrast abdomen MRI.
Source: Document obtained during the study.
The patient was considered to have neoplastic involvement originating in the right adrenal gland with described metastatic lesions. Androgen elevation was found upon completion of the patient’s hormonal profile (Table 2).
Table 2. Hormonal profile of the patient on admission.
|
Test |
Result |
Normal values |
|
Urine metanephrines |
0.32 mg/24 hours |
0-1 mg/24 hours |
|
Androstenedione |
>10 ng/mL |
0.8-2.6 ng/mL |
|
Dehydroepiandrosterone sulfate |
>1000 ug/dL |
44.3-331 ug/dL |
|
Cortisol AM |
28.8 ug/dL |
3.7-19.4 ug/dL |
|
Cortisol PM |
22.4 ug/dL |
2.9-17.3 ug/dL |
|
TSH |
2.29 mUI/L |
0.35-4.94 mUI/L |
TSH: thyroid-stimulating hormone.
Source: Own elaboration.
A biopsy was performed on one of the liver lesions, revealing findings compatible with adrenal cortex tumor and extensive necrosis. Calretinin, melan-A and cytokeratin AE1-AE3markers were positive, with negative CD117 and chromogranin (Figure 3).
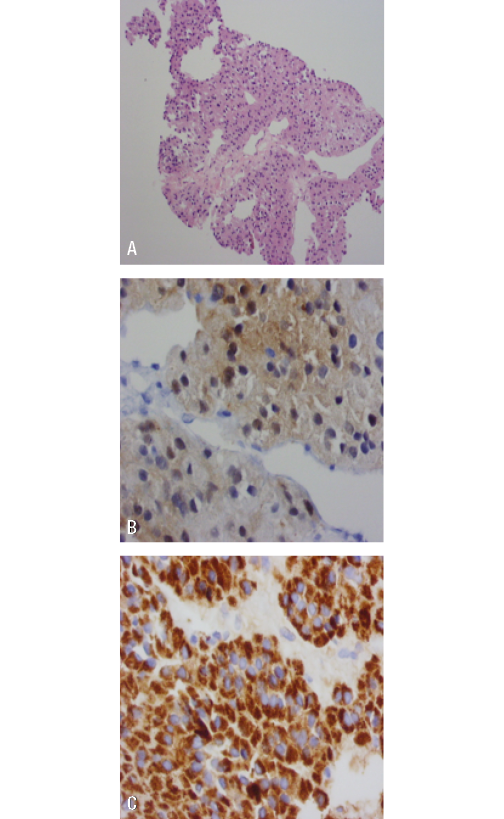
Figure 3. Liver lesion biopsy. A) adrenal gland with HE 40X staining;
B) positive calretinin staining; C) melan-A.
Source: Document obtained during the study.
The patient was taken to a cancer board. Considering the poor prognosis and the contraindication for the use of anthracycline and platinum due to risk of cardiac toxicity, as well as the deleterious effect on the ejection fraction already involved, the patient decided to continue palliative management for heart failure. He was discharged after compensation.
Discussion
Genetic mechanisms
The signals involved in the development of the ACC are associated with the signaling pathway that mediates progenitor cell migration and their differentiation into glomerular and fascicular cells. (4) The over-expressed factors in patients with ACC are fibroblast growth factor receptor 1 and 4 —which have also been associated with worse prognosis—, insulin-like growth factor, and dysfunction of the Wnt signaling pathway. (4)
Clinical presentation
Clinical presentation varies according to the functionality of the carcinoma: although most are biochemically functional, many are diagnosed incidentally or due to metastatic disease. The most common sites of metastasis are liver, lung and bone. (5)
Imaging findings
Characteristics suggesting malignant adrenal lesion in CT scan are irregular margins, heterogeneous density with ring enhancement, and presence of extensive areas of necrosis. (6)
In the study by Petersenn et al. (7), the CT scans of 51 patients were compared with ACC, of which 25 showed evidence of benign adrenal lesions. It was found that size >3.9cm had 98% sensitivity (S), 40% specificity (E), areas under the curves (AUC) of 0.93, and confidence interval (95%CI) of 0.88-0.99 for diagnosis. The non-homogeneous appearance for ACC had OR: 130, S: 100% and E: 56%. The median enhancement in Hounsfield units (UH) was 34, and the value of 21 UH had S: 96%, E: 80%, AUC: 0.89, and 95%CI: 0.79-0.98.
The MRI showed heterogeneous signal on T1 and T2 secondary to the hemorrhage and necrosis areas in the tumor. (6) In the case presented here, the abdominal magnetic resonance showed a lesion of 19x18cm, which was highly suggestive of malignant neoplasm, as well as heterogeneous signal on T1 and T2, which together with the lesions observed in lung and liver, led to the diagnosis of tumor recurrence by ACC.
Functional profile
ACC may be a hormone producer in 76% of patients; however secondary symptoms may occur in only 70% of this group. (8) The most produced hormones are cortisol and androgens, being more frequent their simultaneous secretion (47%). Cortisol is secreted in isolation in 27% of patients and androgens in 6%. Aldosterone or estradiol are observed with a lower frequency. (8)
Endocrine assessment is required prior to any surgical intervention and pheochromocytoma should always be ruled out. Cortisol secretion is associated with risk of post-operative adrenal insufficiency, while elevation of steroid precursors is associated with the possibility of malignancy. Follow-up also requires continued hormone measurement to evaluate recurrences. (9) Table 3 summarizes the tests to be performed for the functional evaluation of adrenal tumors.
Table 3. Hormonal evaluation of adrenal tumors.
|
Excessive function |
Test |
|
Glucocorticoids |
At least 3 of the following tests are recommended: Dexamethasone suppression, cortisol urine tests, basal serum cortisol, or basal plasma ACTH |
|
Steroids and sex steroids |
Measurement of DHEA, 17-OH progesterone, testosterone, 17β-estradiol |
|
Mineralocorticoids |
In case of hypertension or hypkalemia, perform aldosterone-to-renin ratio |
|
Pheochromocytoma |
Urine catecholamines and plasma metanephrines |
Source: Own elaboration based on Allolio & Fassnacht (9).
Histology
To histologically differentiate adenoma from cortical carcinoma, the World Health Organization adopted the Weiss score. With it, malignancy is diagnosed when at least three criteria are met, including high nuclear grade, high mitotic rate (>5 mitoses per 50 high-power fields), atypical mitotic figures, less than 35% clear cells, altered architecture, tumor necrosis, venous invasion, sinusoidal invasion, and capsular invasion. (3) The positive immunohistochemical markers in ACC are steroidogenic factor 1, inhibin alpha, melan-A and calretinin. (3)
Surgical management
Surgical management with open or laparoscopic surgery is recommended for adrenal masses at high risk of malignancy, such as those with secretion or precursors of sex hormones and high uptake in fluorodeoxyglucose positron emission tomography (FDG-PET). The recommended approach is laparoscopic, but if the tumor is larger than 6cm, open surgery is recommended (10) to avoid bursting the capsule and spreading tumor cells. For this reason, the guidelines of the European Society of Endocrine Surgeons recommend block resection and locoregional lymphadenectomy. (10)
In the reported case, the patient initially presented an adrenal mass of 8cm, reason why he was referred to another institution for adrenalectomy by laparoscopy, which ultimately led to the development of the recurrence. One of the limitations of this report is that the surgical report was not available to describe the intraoperative findings. Intraoperative rupture and positive resection margins (R2) are associated with higher rates of recurrence and lower survival (10), so the lack of knowledge of these two factors —given the lack of surgical assessment— made it difficult to establish the initial prognosis for the patient.
Post-operative follow-up
After surgery, clinical, hormonal and imaging evaluation should be performed with CT scans and FDG-PET every 3 months for 2 years, and then every 4 to 6 months. (10) This follow-up was not made on the patient, resulting in the late diagnosis of neoplastic recurrence.
Adjuvant cancer treatment
Adjuvant cancer treatment for patients at high risk of recurrence includes the use of mitotane, which has shown benefits both in monotherapy and in combination with other cytotoxics. (11)
Although the mechanism is not clear, it is assumed that the metabolites formed after mitotane oxidation bind to mitochondrial proteins that inhibit the respiratory chain and enzymes of steroid biosynthesis. (11) The combination of mitotane with etoposide, doxorubicin and cisplastin has been suggested as the adjuvant therapy of choice for the treatment of adrenal carcinoma in patients at high risk of recurrence or late stage. (12) Its use as second-line treatment has even shown a median progression-free survival of 5.6 months in patients who received etoposide, doxorubucin, and cistplatin plus mitotane versus 2.2 months in patients who received second-line streptozocine plus mitotane. (12)
In this case, considering the poor prognosis and the contraindication for anthracycline and platinum due to cardiac toxicity, the patient made the decision to receive palliative therapy.
Tumor recurrence
The 5-year survival rate depends on the initial tumor stage; thus, survival is 82% for stage I, 61% for stage II, 50% for stage III, and 13% for stage IV. (10)
Factors associated with recurrence are tumor >12cm, positive lymph nodes, stages III and IV, cortisol secretion, capsular invasion (13), Ki-67 >10%, and tumor spread during surgery. (10) Recurrence time is 1.2 to 2.5 years, with a median of approximately 1 year between resection and first recurrence. (14-16)
Since late recurrence is rare, the longest time interval between initial treatment and recurrence is 30 years. (17) Diagnosis can be made incidentally (18), either based on clinical findings suggestive of hormonal secretion (19,20), or signs of metastatic involvement, where skin metastasis manifested as a dorsal cystic lesion is the initial sign of recurrence. (17)
Locoregional recurrence may occur in 20-60% of patients. (10) At the adrenalectomy site, as in this case, it occurs in up to 41% of the cases of patients with isolated recurrence. (21) Other sites of isolated recurrence, in order of frequency, are liver, lung and brain. (21)
In cases of recurrence, the indicated management is open surgery; however, if reoperation is determined, the risk of morbidity and mortality must be taken into account. (10) The use of mitotane should be considered as an adjuvant treatment. (10,22)
Management of metastatic disease includes, in addition to surgery, options such as radiation therapy. In symptomatic metastatic lesions, the response to pain improvement occurs in 72% of cases. (22) In patients with limited life expectancy, the decision to receive any type of therapy should be made together with the patient, taking into account the risk of cardiac toxicity and survival rates; in the present case, the patient made the decision not to receive any type of therapy.
Conclusions
The reported case is an unfortunate outcome resulting from the lack of strict follow-up in a patient with a history of ACC, since multiple recurrences could be detected earlier. Therefore, adherence to follow-up is important, as recommended by the guidelines, to detect recurrences in a timely manner and have interventions that improve outcomes.
The high mortality associated with stages III and IV is the reason to make further research on adjuvant treatment in ACC with other cytotoxics, IGF1 and radiotherapy. To date, the only scheme established is the combination of mitotane with etoposide, doxorubicin and cisplastin, which could not be used in the patient due to concomitant heart failure.
Ethical considerations
This article was approved by the Research Ethics Committee of the Fundación Cardioinfantil - Instituto de Cardiología, as stated in Minutes 20-2018 of June 20, 2018. This report ensures the privacy of the patient.
Conflicts of interest
None stated by the authors.
Funding
None stated by the authors.
Acknowledgements
None stated by the authors.
References
1.Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, et al. Adrenocortical Carcinoma. Endocr Rev. 2014;35(2):282-326. http://doi.org/f5zwss.
2.Kebebew E, Reiff E, Duh QY, Clark OH, McMillan A. Extent of disease at presentation and outcome for adrenocortical carcinoma: havewe made progress? World J Surg. 2006;30(5):872-8. http://doi.org/b6m3rj.
3.Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. EndocrPathol. 2017;28(3):213-27. http://doi.org/c345.
4.Åkerström T, Carling T, Beuschlein F, Hellman P. Genetics of adrenocortical tumours. J InternMed. 2016;280(6):540-50.http://doi.org/f3td3s.
5.Wanis KN, Kanthan R. Diagnostic and prognostic features inadrenocortical carcinoma : a single institution case series and review of the literature. World J SurgOncol. 2015;13:117. http://doi.org/f67ss6.
6.FarrugiaFa, Martikos G, Surgeon C, Tzanetis P, Misiakos E, Zavras N, et al. Radiology of the adrenal incidentalomas. Review of the literature. EndocrRegul. 2017;51(1):35-51. http://doi.org/c346.
7.Petersenn S, Richter PA, Broemel T, Ritter CO, Deutschbein T, Beil FU, et al. Computed tomography criteria for discrimination of adrenal adenomas and adrenocortical carcinomas : analysis of the German ACC registry. Eur J Endocrinol. 2015;172(4):415-22. http://doi.org/f636pw.
8.Abiven G, Coste J, Groussin L, Anract P, Tissier F, Legmann P, et al. Clinical and biological features in the prognosis of adrenocortical cancer: pooroutcome of cortisol-secreting tumors in a series of 202 consecutive patients. J ClinEndocrinolMetab. 2006;91(7):2650-5. http://doi.org/c9szw8.
9.Allolio B,Fassnacht M. Adrenocortical Carcinoma: ClinicalUpdate. J ClinEndocrinolMetab. 2006;91(6):2027-37. http://doi.org/dkfdfg.
10.Gaujoux S, Mihai R. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma. Br J Surg. 2017;104(4):358-76.http://doi.org/f9qbjt.
11.Ronchi CL, Kroiss M, Sbiera S, Deutschbein T, Fassnacht M. EJE PRIZE 2014: Current and evolving treatment options in adrenocortical carcinoma: where do we stand and where do we want to go? Eur J Endocrinol. 2014;171(1):R1-11. http://doi.org/f582sw.
12.Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. Combination Chemotherapy in Advanced Adrenocortical Carcinoma. N Engl J Med. 2012;366(23):2189-97. http://doi.org/f228dm.
13.Kim Y, Margonis GA, Prescott JD, Tran TB, Postlewait LM, Maithel SK, et al. Nomograms to Predict Recurrence-Free and Overall Survival After Curative Resection of Adrenocortical Carcinoma. JAMA Surg. 2016;151(4):365-73. http://doi.org/c347.
14.Dy BM, Wise KB, Richards ML, Young WFJr, Grant CS, Bible KC, et al. Operative intervention for recurrent adrenocortical cancer. Surgery. 2013;154(6):1292-9. http://doi.org/f5h4n2.
15.Erdogan I, Deutschbein T, Jurowich C, Kroiss M, Ronchi C, Quinkler M, et al. The role of surgery in the management of recurrent adrenocortical carcinoma. J ClinEndocrinolMetab. 2013;98(1):181-91. http://doi.org/f4kjdw.
16.Icard P, Chapuis Y, Andreassian B, Bernard A, Proye C. Adrenocortical carcinoma in surgically treated patients: a retrospective study on 156 cases by the French Association of Endocrine Surgery. Surgery. 1992;112(6):972-80.
17.Satter EK, Barnette DJ. Adrenocortical carcinoma with delayed cutaneous metastasis. J Cutan Pathol. 2008;35(7):677-80. http://doi.org/bkg6dc.
18.Mawardi M, Al-Judaibi B, Marotta P. Hepatic metastasis from adrenocortical carcinoma fifteen years after primary resection. Saudi J Gastroenterol. 2012;18(2):140-2. http://doi.org/c348.
19.Iino K, Oki Y, Sasano H. A case of adrenocortical carcinoma associated with recurrence after laparoscopicsurgery. ClinEndocrinol (Oxf). 2000;53(2):243-8. http://doi.org/dx7z6z.
20.Orlando R, Pelizzo MR, Lirussi F. Adrenocortical carcinoma: a 15-year survival after complete resection and repeated resection. A retrospective study in a patient with an expected poor prognosis. Anticancer Res. 2003;23(3C):2929-31.
21.Simon G,Pattou F,Mirallié E,Lifante JC,Nominé C,Arnault V,et al. Surgeryforrecurrentadrenocortical carcinoma : A multicenterretrospectivestudy. Surgery. 2017;161(1):249-56.http://doi.org/f9hdwp.
22.Fay AP, Elfiky A, Teló GH, McKay RR, Kaymakcalan M, Nguyen PL, et al. Adrenocortical carcinoma: the management of metastatic disease. Crit Rev Oncol Hematol. 2014;92(2):123-32. http://doi.org/c5ff.
Jean-Baptiste Marc Bourgery. (1797- 1849)
“Traité complet de l’anatomie de l’homme: comprenant la médecine operatoire”