Red de Revistas Científicas de América Latina y el Caribe, España y Portugal
Aplicada y Analítica
Método cualitativo rápido (screening) para la detección de residuos de plaguicidas en frutas y hortalizas
Qualitative screening method for pesticide residues detection in fruits and vegetables
Método qualitativo rápido (screening) para a detecção de resíduos de pesticidas nas frutas e nos vegetais
Método cualitativo rápido (screening) para la detección de residuos de plaguicidas en frutas y hortalizas
Revista Colombiana de Química, vol. 47, núm. 1, 2018
Universidad Nacional de Colombia
Recepción: 08 Febrero 2017
Aprobación: 25 Julio 2017
Resumen: Debido a la importancia de desarrollar metodologías que permitan el análisis de los residuos agrícolas, el presente trabajo validó un método cualitativo rápido (screening) para el análisis de residuos de plaguicidas en frutas y hortalizas. La metodología se basó en el método de extracción QuEChERS, versión europea, con un paso adicional de limpieza por cromatografía de permeación por gel (GPC), lo cual permitió reducir la cantidad de componentes de la matriz en el extracto final. El análisis fue realizado por cromatografía de gases/espectrometría de masas con un analizador cuadrupolo simple. La metodología resultó adecuada para el análisis cualitativo de 31 plaguicidas a su respectivo límite máximo de residuos. Los resultados en muestras reales fueron consistentes respecto a una metodología cuantitativa de rutina, por ende, la metodología resultó ser una buena alternativa para el análisis rápido de estos contaminantes.
Palabras clave: cromatografía de gases, espectrometría de masas, QuEChERS, validación, método cualitativo rápido.
Abstract: Because of the importance of developing methodologies that allow agricultural residues analysis, a rapid screening qualitative method for the determination of pesticides residues in fruits and vegetables was validated. The methodology was based on the European QuEChERS extraction method with an additional cleaning step by gel permeation chromatography (GPC), which helped to reduce the number of matrix components in the final extract. The analysis was carried out by gas chromatography coupled to mass spectrometry with a single quadrupole analyzer. The methodology was appropriate for the qualitative analysis of 31 pesticides at their respective maximum residue limits. Consistent results were obtained with respect to a quantitative routine methodology in the analysis of real samples, hence the methodology was proven to be a good alternative for the fast analysis of these contaminants in fruits and vegetables.
Keywords: Gas chromatography, mass spectrometry, QuEChERS, validation, screening method.
Resumo: Devido à importância de desenvolver metodologias que permitam analisar os resíduos agrícolas, o presente trabalho validou de um método qualitativo rápido (screening) para a análise de resíduos de pesticidas nas frutas e vegetais. A metodologia foi baseada no método de extração QuEChERS versão Europeia com um passo adicional de limpeza por meio de cromatografia de permeação em gel (GPC), o que permitiu reduzir a quantidade de componentes de matriz no extrato final. A análise foi realizada por cromatografia gasosa/espectrometria de massa com um analisador de quadrupolo simples. A metodologia foi adequada para a análise qualitativa de 31 pesticidas aos seus respectivos limites máximos de resíduos. Os resultados obtidos em amostras reais foram consistentes com uma metodologia quantitativa de rotina, portanto, a metodologia estudada tem demonstrado ser uma boa alternativa para a análise rápida destes contaminantes em frutas e vegetais.
Palavras-chave: cromatografia em fase gasosa, espectrometria de massa, QuEChERS, validação, método de triagem.
Introducción
La protección de cultivos juega un papel importante en la producción de alimentos a nivel mundial. Una de las formas de protección más utilizada en la agricultura ha sido el uso de plaguicidas, en respuesta a la creciente demanda de alimentos, al incremento constante de la población y a la limitada área de terreno cultivable disponible. El uso de este tipo de agroquímicos ha permitido incrementar la producción de frutas, hortalizas, cereales, etc. y, del mismo modo, mejorar la calidad de los productos que son comercializados a nivel mundial (1).
Con el fin de garantizar la seguridad en el consumo de alimentos y facilitar la comercialización, organismos internacionales han establecido Límites Máximos de Residuos (LMR) de plaguicidas en alimentos. La Comisión del Codex Alimentarius (CAC) de la Organización de las Naciones Unidas para la Alimentación y la Agricultura (FAO) en conjunto con la Organización Mundial de la Salud (WHO) (2), la unión europea (EU) (3), la Agencia de Protección ambiental de los Estados Unidos (US-EPA) (4), el Centro de Inspección de Materiales de Agricultura y Alimentos (FAMIC, Japón) (5), entre otros, han utilizado los LMR con el propósito de llevar un registro, monitoreo y control de los residuos de plaguicidas en un amplio rango de alimentos, tanto de origen vegetal como de origen animal.
Ahora bien, debido a que un número mayor a 1000 sustancias activas están siendo actualmente utilizadas en el control de plagas a nivel mundial (6), los laboratorios han tenido la necesidad de desarrollar métodos multiresiduo con el fin de ejercer un control efectivo que permita la determinación de un gran número de compuestos en un solo análisis (7). Así, en los últimos años, se han implementado métodos multiresiduo cuantitativos basados en cromatografía liquida (HPLC) (8), cromatografía de gases (9, 10, 11) y cromatografía de gases acoplada a espectrometría de masas (GC-MS) (12).
El uso de este tipo de metodologías requiere la construcción de curvas de calibración en matriz, realizar ensayos de recuperación, determinar dispersión de los resultados debido al gran número de plaguicidas trabajados. Lo anterior requiere bastante tiempo de análisis y un alto número de datos a procesar, por lo cual recientemente se han implementado métodos cualitativos rápidos (screening). Estos métodos están enfocados principalmente en determinar la presencia o ausencia de un compuesto en una muestra de ensayo a una concentración superior del límite establecido (por lo general el LMR) sin necesidad de cuantificar. La principal ventaja en el uso de este tipo de metodologías es la posibilidad de realizar una identificación rápida de los posibles residuos presentes en una muestra antes del uso de un método cuantitativo de rutina, lo que permite la selección de posibles muestras contaminadas, reduciendo la carga de trabajo y el costo en un análisis de cuantificación y confirmación (13).
Estos métodos screening se han venido utilizando en análisis de rutina, empleando cromatografía liquida y cromatografía de gases acoplada a espectrometría de masas de alta resolución (GC(LC)-HRMS) en modalidad Full Scan. Este tipo de configuración es la ideal para la detección simultánea de un gran número de compuestos y brinda una gran confiabilidad en la identificación gracias a la determinación de las masas exactas.
Múltiples trabajos con el uso de espectrometría de masas de alta resolución han sido publicados en los últimos años (14, 15, 16, 17, 18). Entre estos se pueden destacar: Portoles et al. (13), quienes desarrollaron un método screening para el análisis de 150 contaminantes orgánicos en agua empleando cromatografía de gases acoplada a espectrometría de masas con un analizador de tiempo de vuelo (GC-TOF MS), obteniendo límites de detección entre 0,02 a 1 µg/mL. Este trabajo demostró la aplicación de los métodos screening en la identificación de contaminantes de importancia en el área ambiental. García et al. (17) desarrollaron un método para el análisis de 199 plaguicidas en frutas y hortalizas empleando LC-TOF MS, donde se obtuvieron límites de detección entre 0,01 y 0,05 mg/kg, la optimización de los parámetros de adquisición del método permitió el procesamiento automático de los datos facilitando la implementación de los métodos screening. En Rajski et al. (18) se desarrolló un método para el análisis de 170 plaguicidas en frutas y hortalizas empleando LC-Orbitrap (Cromatografía liquida-trampa orbital) obteniendo límites de detección entre 0,01 y 0,05 mg/kg. Este trabajo demostró la capacidad de la instrumentación y el enfoque de los métodos screening para la identificación de un gran número de compuestos en frutas y hortalizas.
En su mayoría, los métodos screening publicados se basan en el uso de instrumentación avanzada como la espectrometría de masas de alta resolución. Muy pocos trabajos han sido publicados con el uso de espectrometría de masas de baja resolución (19, 20), de uso común en los laboratorios de América Latina. En Mol et al. (20) se desarrolló un método para el análisis de 94 plaguicidas en frutas y hortalizas empleando cromatografía de gases acoplada a espectrometría de masas con un analizador de cuádruplo simple GC-MSD, inyección de grandes volúmenes con un inyector de temperatura de vaporización programada (PTV) y adquisición en el modo Full Scan empleando un software de deconvolución, obteniendo como resultado la detección del 82% de los compuestos a una concentración de 0,01 mg/kg.
Por lo anterior, siendo Colombia un país con gran potencial de exportación, es importante la implementación y el desarrollo de metodologías analíticas con la capacidad de determinar residuos de plaguicidas a bajas concentraciones, con el fin de garantizar que sus productos cumplan con la normatividad. Por esta razón el objetivo del presente trabajo consistió en el montaje y posterior validación de un método screening para el análisis de 52 plaguicidas en frutas y vegetales con alto contenido de agua empleando cromatografía de gases acoplada a espectrometría de masas con cuádruplo simple (GC-MSD). Una vez se validó el método se analizaron frutas y hortalizas para evaluar la efectividad de la metodología. Los resultados se compararon con un método cuantitativo de rutina, validado y acreditado bajo la norma ISO 17025 (21).
Materiales y métodos
Patrones de referencia, reactivos y soluciones
Se emplearon estándares de plaguicidas con una pureza mayor al 95%, se adquirieron de la casa comercial Dr. Ehrenstorfer (Augsburg, Alemania). La Tabla 1 muestra los plaguicidas incluidos en el estudio. Soluciones madre se prepararon a una concentración cercana a 1000 µg/mL en acetato de etilo y se almacenaron en frascos ámbar a -20 °C. Una solución patrón, mezcla de plaguicidas, se preparó tomando diferentes volúmenes de cada solución madre hasta obtener una concentración de cada plaguicida cercana a 1000 ng/mL. Esta solución se almacenó en un frasco ámbar a -20 °C. Se empleó acetato de etilo grado residuos, ciclohexano y acetonitrilo grado HPLC de J.T. Baker (USA). El sulfato de magnesio, citrato di-sódico sesqui hidratado y citrato tri-sódico di hidratado se adquirieron de Sigma Aldrich (Alemania); el cloruro de sodio se adquirió de J.T. Baker (USA) y la amina primaria sedundaria, PSA (N-propiletilendiamina) se adquirió de Agilent Technologies (USA).

Instrumentos y equipos
En el proceso de extracción se empleó un homogeneizador Stephan Blender 2010 (Hamein, Alemania) y una licuadora industrial (Warring), para el proceso de limpieza se utilizó un cromatógrafo de permeación por gel de Redemnt Bt, modelo KL-SX-3, equipado con una columna de vidrio de 10 cm x 10 mm de diámetro interno. Como fase estacionaria se usó un gel Biobeads S-X3, y como fase móvil se empleó una mezcla de acetato de etilo: ciclohexano (1:1) a un flujo de 1 mL/min.
Para el análisis del método screening se empleó un cromatógrafo de gases Agilent Technologies Modelo 7890A acoplado a un detector selectivo de masas (MSD) 5975C, una columna capilar TBR-5MS (30 m x 0,25 mm d.i x 0,25 µm espesor). Las condiciones cromatográficas utilizadas en el análisis fueron las siguientes: volumen de inyección 4 µL; inyección en modalidad splitless pulsado (22) con presión de pulso de 65 psi durante 0,8 min; tiempo de purga de 0,8 min y temperatura del inyector de 250 °C. El gas de transporte fue helio en modo presión constante a 19 psi. La temperatura de la línea de transferencia al espectrómetro de masas fue 280 °C y la temperatura del cuadrupolo fue de 150 °C. El programa de temperatura del horno inició a 50 °C (5 min), se incrementó a una velocidad de 20 °C/min hasta alcanzar 195 °C; esta temperatura se mantuvo por 5 min. Después, se incrementó la temperatura hasta 280 °C a 10 °C/min; esta temperatura se mantuvo por un tiempo de 7,5 min.
El modo de ionización fue por impacto electrónico a 70 eV, con una temperatura de la fuente de ionización de 230 °C. En el análisis se empleó monitoreo de ion selectivo (SIM). La selección de los iones se realizó inyectando una solución con los compuestos de interés a una concentración aproximada de 500 µg/mL bajo el modo de adquisición Full Scan. Se seleccionaron cuatro iones por compuesto a partir del espectro de masas, correspondientes a un ion objetivo y 3 iones de confirmación o cualificadores. Los parámetros de adquisición se muestran en la Tabla 1.
En el análisis cuantitativo de rutina se empleó un cromatógrafo de gases Agilent Technologies HP6890 plus con un inyector split/splitless, el cual se conectó a través de una columna capilar sin fase estacionaria (1 m x 0,32 mm d.i) a un divisor de flujo de cuarzo en forma de “Y”, unido a una columna capilar HP-5 (30 m x 0,32 mm d.i.x 0,25 µm) acoplada a un detector de micro-captura electrónica (µ-ECD) y a una columna capilar HP-50 (30 m x 0,32 mm d.i. x 0,25 µm) acoplada a un detector de nitrógeno-fosforo (NPD) en paralelo. Las condiciones de operación fueron las siguientes: gas de transporte nitrógeno, volumen de inyección 2 µL, temperatura del inyector 256°C, inyección en modo splitless pulsado (22) con presión de pulso de 65 psi durante 0.8 min, tiempo de purga 0.6 min y flujo de purga de 40 mL/min. La temperatura del detector µ-ECD fue de 310°C con flujo de gas auxiliar (nitrógeno) 10 mL/min. El detector NPD se trabajó a 330°C con flujo constante de gas auxiliar (nitrógeno), hidrógeno y aire de 10, 3 y 60 mL/min, respectivamente. El programa de temperatura del horno inició a 52°C (0 min), incrementándose la temperatura a una velocidad de 4°C/min hasta alcanzar 100°C, consecutivamente se aumentó la temperatura hasta 110°C a 2°C/min, a continuación, se incrementó hasta 130°C a una velocidad de 20°C/min, luego se llevó hasta 195°C a 4°C/min y finalmente se llegó a una temperatura de 280°C a una velocidad de 5°C/min.
Método de preparación de muestra
Las frutas y vegetales frescos empleados se compraron en supermercados; se transportaron y procesaron el mismo día. La preparación del producto se llevó acabo tal como es descrito en el Codex Alimentarius (23). Alrededor de 2 kg de cada producto se homogeneizó en el homogeneizador por 5 min; porciones de muestra procesada (100 g) se almacenaron en bolsas herméticas a -20 °C hasta el análisis.
El método empleado corresponde a una modificación del proceso de extracción QuEChERS versión europea (24). Se tomaron 10 g de muestra homogeneizada en un tubo de polipropileno de 50 mL; se agregaron 30 µL de estándar subrogado y, luego, se adicionaron 10 mL de acetonitrilo grado HPLC. Se realizó una agitación manual durante 1 min; luego se adicionó una mezcla de sales compuesta de 4 g de MgSO4 anhidro, 1 g de NaCl, 1 g de citrato tri sódico di hidratado y 0,5 g de citrato di sódico sesqui hidratado. La mezcla se agitó manualmente por 2 min y se centrifugó a 4500 rpm por 5 min a 10 °C. Se observaron cuatro fases en los tubos de centrifugación: una fase superior orgánica donde se encuentran los plaguicidas a analizar; una fase intermedia correspondiente a la matriz; una fase acuosa y, por último, una que contiene las sales sólidas.
Posteriormente se colocaron 2 mL de fase orgánica en un tubo de polipropileno de 15 mL, se realizó una limpieza por extracción en fase sólida dispersiva (d-SPE) con 290 mg de MgSO4 anhidro y 50 mg de PSA, empleando un agitador vortex por 2 min. El tubo se centrifugó a 4500 rpm por 5 min a 10 °C. Se tomó 1 mL de sobrenadante y se llevó a sequedad en un evaporador rotatorio a 35 °C, el extracto se transfirió cuantitativamente a un balón aforado de 1 mL completando volumen con acetato de etilo.
Seguidamente, se inyectaron 500 µL de extracto en el cromatógrafo de permeación por gel. La fracción de los plaguicidas (7–22 mL) se recolectó en un balón en forma pera de 20 mL. El extracto se concentró en un evaporador rotatorio a 35 °C; se adicionaron 10 µL de estándar interno y 490 µL de acetato de etilo. El extracto se homogeneizó en un agitador vortex por 1 min. Por último, el extracto se transfirió a un vial y se inyectó en el cromatógrafo de gases acoplado a espectrometría de masas.
Protocolo de la validación cualitativa
La validación del método screening se basó principalmente en el documento SANTE/EU 2015 (25) y en la directiva de la comisión europea EU 657/2002 (26). Los parámetros de validación respecto a estos documentos corresponden a selectividad, especificidad, límite de detección del método screening (SDL) y aplicabilidad. Para la validación fueron seleccionados diez productos diferentes representativos del grupo de frutas y hortalizas con alto contenido de agua (piña, granadilla, banano, uchuva, pitaya, papa, naranja, repollo, lechuga y pepino).
La selectividad, definida como la habilidad de un método en discriminar entre la señal del analito y cada una de las demás señales, y la especificidad, definida como la habilidad del detector (soportada por la selectividad del método de extracción, limpieza y separación) en proveer señales que efectivamente identifican al analito (25), se evaluaron inyectando una solución de la mezcla de analitos en extracto de matriz a una concentración de 100 ng/mL y dos muestras de cada producto sin fortificar, para un total de 20 ensayos.
Se determinó el tiempo de retención de cada analito con una tolerancia de ± 0,1 min. Así mismo, se identificaron interferencias de cada matriz y cuatro iones característicos para cada compuesto como se muestra en la Tabla 1.
Para la evaluación del límite de detección del método screening (SDL) se realizó el análisis de dos muestras de cada producto, las cuales se fortificaron con la mezcla de plaguicidas a 10 µg/kg, la concentración más baja permitida como límite máximo de residuos de plaguicidas en frutas y hortalizas. También se fortificaron a 30 y 100 µg/kg, debido a que hay compuestos cuya sensibilidad no es suficiente para detectar la concentración de 10 µg/kg por GC/MS. Adicionalmente, hay plaguicidas que tienen límites máximos de residuos superiores en este tipo de matrices. Se analizó para cada concentración un total de 20 ensayos, 10 muestras por duplicado, para un total de 60 análisis.
Para la determinación del SDL se estableció como criterio de identificación la presencia del ion objetivo y por lo menos dos iones cualificadores al tiempo de retención de cada compuesto (al menos tres iones seleccionados del cromatograma) y el cumplimento de las relaciones de intensidad entre el ion objetivo (Q) y cada ion de confirmación (qi) (relaciones de abundancia) como lo sugiere el documento SANTE/EU 2015 (25). El SDL se estableció como la concentración más baja en la que el compuesto de interés se detectó satisfactoriamente en un 95% de los 20 ensayos (detección en un mínimo de 19 muestras a cada nivel de concentración evaluado) independiente de su porcentaje de recuperación y coeficiente de variación (25).
En la evaluación de la aplicabilidad, se analizaron muestras correspondientes a lulo, lechuga, zanahoria, brócoli, coliflor y uchuva empleando la metodología descrita. Los resultados se compararon con una metodología cuantitativa de rutina validada y acreditada bajo la norma ISO 17025 (21).
Resultados y discusión
Especificidad – Selectividad
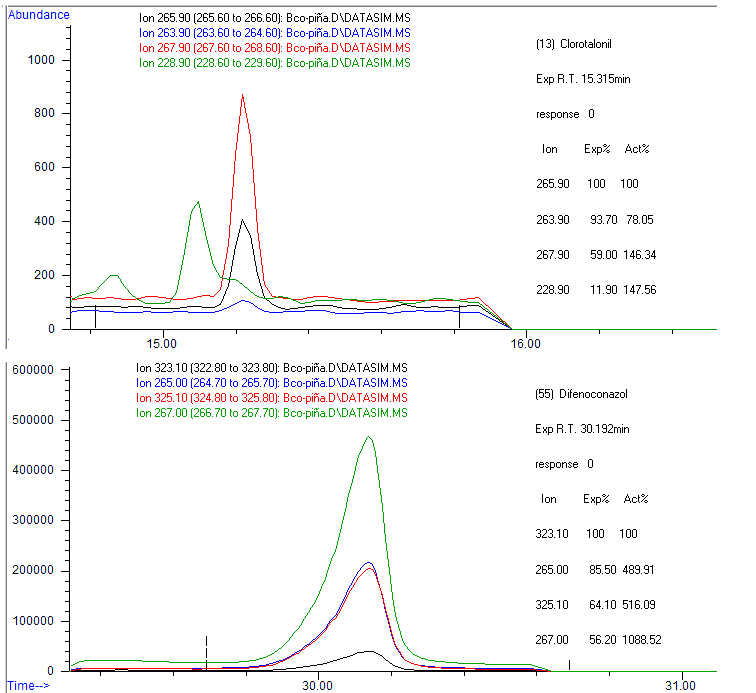
En la evaluación de la especificidad-selectividad de la metodología, se evidenció una correcta separación cromatográfica para la mayoría de los analitos (resolución mayor a 1,5). Compuestos como el pp-DDE, dieldrin, propiconazol, pp-DDT presentaron una resolución menor a 1,0 y fueron correctamente detectados por espectrometría de masas gracias a la selección de los iones característicos para cada analito. Se encontraron señales en los tiempos de retención de los compuestos clorotalonilo, α-Endosulfan, propargite y difenoconazol que impidieron su correcta identificación.
La Figura 1 muestra los iones que se seleccionaron para clorotalonilo y difinoconazol, así como sus abundancias esperadas (Exp%) y experimentales obtenidas en el análisis (Act%). Para el clorotalonilo se observa que las abundancias esperadas y experimentales son muy diferentes para los iones cualificadores 263,9; 267,9 y 228,9. Lo mismo sucede para los iones 265,0; 325,1 y 267,0 en el difenoconazol. Por tanto, estos compuestos no cumplen los criterios de identificación, pues las relaciones esperadas y experimentales son muy diferentes (26). Estas señales no se pueden atribuir al compuesto de interés, sino a interferentes provenientes de la matriz.
En las muestras de pitaya se encontró la presencia de λ-cihalotrina y en las muestras de pepino se encontró la presencia de difenoconazol a unas concentraciones por debajo de 10 µg/kg, a pesar de esto se incluyeron dentro de la validación pues estos valores tan bajos no interfieren en dicho proceso.
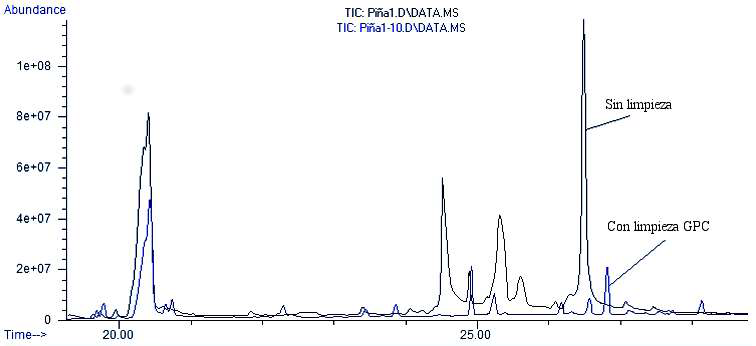
Para mejorar la selectividad del método se empleó un paso de limpieza adicional por cromatografía de permeación por gel (GPC) debido a la gran cantidad de interferencias observadas en los cromatogramas. El proceso de limpieza por GPC permitió reducir en gran medida el número de compuestos de la matriz en el extracto final que interferían con el análisis de los plaguicidas evaluados. En la Figura 2 se muestra la superposición de dos cromatogramas con y sin limpieza adicional, obtenidos en el modo de adquisición Full Scan en matriz de piña sin fortificar. Como se puede observar el número de señales y la intensidad de las mismas es mucho menor en el ensayo realizado con limpieza por GPC. Esta reducción en el número de compuestos de la matriz también permitió mejorar el desempeño instrumental e incrementar el número de muestras a analizar sin realizar un mantenimiento al sistema cromatográfico.
Límite de detección método screening (SDL)
En la determinación del SDL se evaluó la detección de los analitos en cada una de las diez matrices analizadas para cada concentración evaluada por duplicado (10, 30 y 100 µg/kg). El SDL corresponde a la concentración más baja de cada compuesto en por lo menos 19 de 20 muestras fortificadas. Esto corresponde a una aceptación del 5% de probabilidad de falsos negativos (indicación de la ausencia de un analito en una matriz fortificada) como se establece por normatividad (25). Los resultados de la evaluación se muestran en la Tabla 2, donde se presenta el número de veces que es detectado cada plaguicida en los distintos ensayos a las concentraciones evaluadas, el SDL asignado y el LMR.
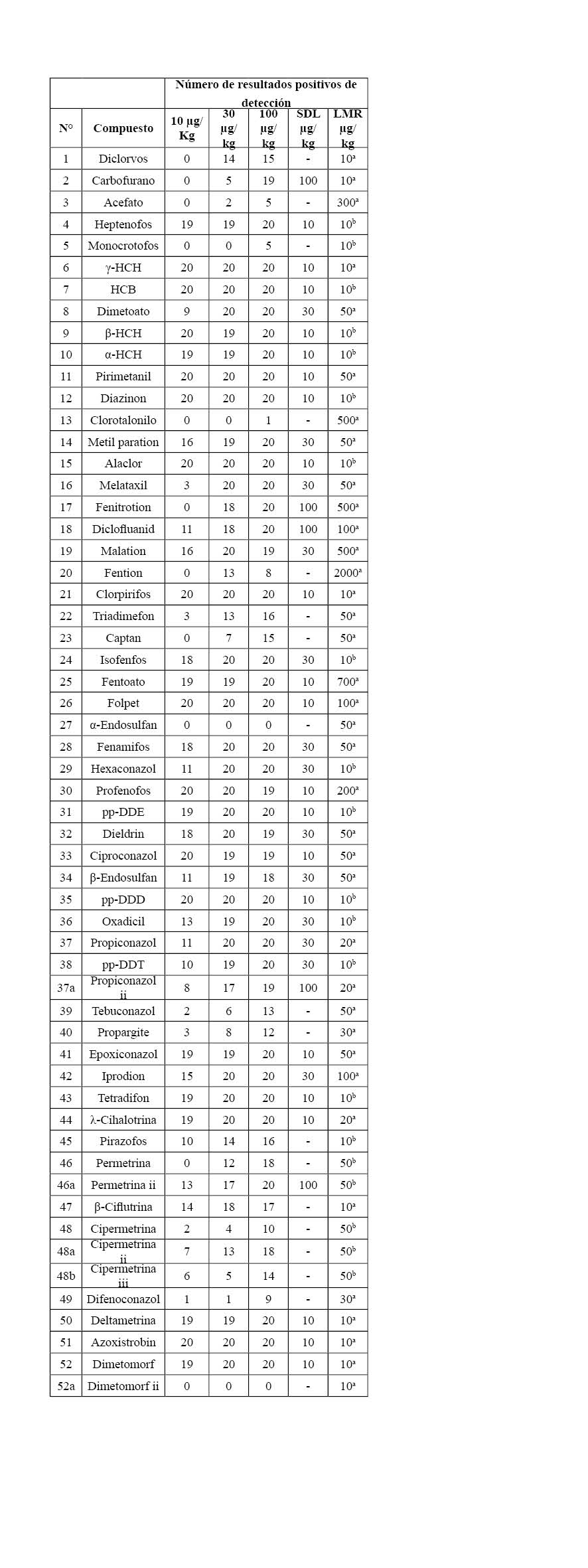
Se asignó como SDL la concentración más baja en la que el plaguicida se detectó en un mínimo de 19 ensayos. Como se muestra en la Tabla 2, a clorpirifos se le asignó un SDL de 10 µg/kg puesto que se detectó 20 veces a las tres concentraciones evaluadas, el valor asignado corresponde a la concentración más baja. En el caso de metalaxil se detectó tres veces a la concentración de 10 µg/kg, 20 veces a 30 µg/kg y 20 veces a 100 µg/kg, por tanto, se le asignó un SDL de 30 µg/kg. En el caso de uno de los isómeros del dimetomorf no se detectó ninguna vez a las tres concentraciones, por tanto, no se le asignó un SDL.
Fue posible asignar un SDL a una concentración igual o inferior al límite máximo de residuos establecido por el Codex Alimentaius (2) y por la comisión europea (3), para un total de 31 compuestos (60%) como se muestra en la Tabla 2. Por tanto, la metodología permite una rápida identificación de estos analitos en una gran variedad de matrices.
Se detectaron con el 95 % de confianza, 21 plaguicidas (40%) a una concentración de 10 µg/kg, 13 (25%) plaguicidas a una concentración de 30 µg/kg y 3 plaguicidas (carbofurano, diclofluanid, y permetrina) a una concentración de 100 µg/kg. Es importante tener en cuenta que plaguicidas como γ-HCH, α-HCH, β-HCH, diazinon, pirimetanil, clorpirifos, folpet, profenofos, pp-DDE, pp-DDD, azoxistrobin, deltametrina y dimetomorf se pueden determinar inclusive a concentraciones por debajo de 10 µg/kg, que es la concentración más baja a detectar en analitos que no tienen un LMR dado, pues la respuesta de estos compuestos fue bastante alta bajo las condiciones de trabajo.
No fue posible establecer un SDL, para los compuestos diclorvos, acefato, monocrotofos, clorotalonilo, fention, triadimefon, captan, α-endosulfan, tebuconazol, propargite, pirazofos, β-ciflutrina, cipermetrina y difenoconazol, inclusive a una concentración tan alta como la de 100 µg/kg, debido a la presencia de interferencias que impidieron su correcta identificación en la mayoría de las matrices que se evaluaron.
En el caso de compuestos como fention, captan, propargite y cipermetrina la sensibilidad de los iones seleccionados fue muy baja, por lo que en varias muestras no fue posible distinguir la señal del analito en la matriz fortificada.
Compuestos como el clorotalonilo, captan y diclofluanid son de particular dificultad en el análisis por el método QuEChERS por su susceptibilidad al pH, a la influencia de la matriz, y a la alta probabilidad de formar productos de degradación durante el proceso de extracción (27). Compuestos como los piretroides presentan una baja sensibilidad por cromatografía de gases/espectrometría de masas, problema ya reportado por otros autores (13).
La Figura 3 muestra el número de plaguicidas detectados a cada concentración evaluada (10, 30, 100 µg/kg) para cada matriz. Como se puede observar el número de plaguicidas detectados a 10 µg/kg fue menor en Uchuva, Naranja, Granadilla y Pepino, indicando que estas matrices son más complejas de analizar. La dificultad en el análisis de plaguicidas en matrices como la naranja ya ha sido reportada por otros autores (28, 29). Lo anterior se genera, principalmente, por la alta cantidad de compuestos inherentes a la matriz que se extraen junto con los analitos de interés y por tanto la alta posibilidad de coelución entre ellos.
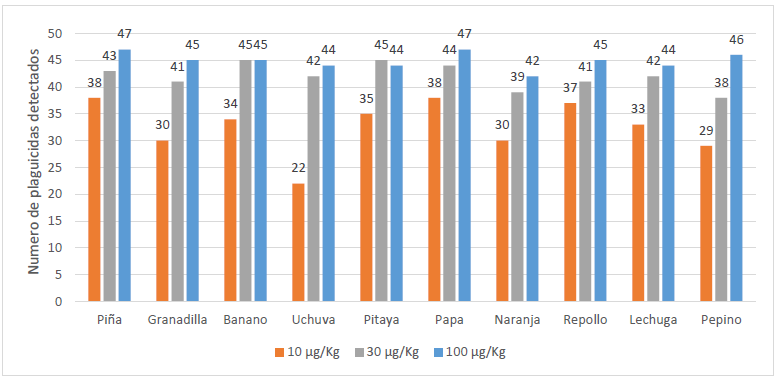
La presencia de compuestos de la matriz con masas similares a los plaguicidas objetivo genera un gran problema en la correcta identificación. Los compuestos isobáricos (igual masa nominal) dificulta el análisis especialmente en equipos de espectrometría de masas de baja resolución, debido a la alta probabilidad de que la instrumentación analítica no pueda resolver estas pequeñas diferencias en masa (29).
Debido a estas matrices, no fue posible asignar un SDL a bajas concentraciones para varios compuestos involucrados en la metodología, pues por las interferencias se dificultó su identificación, disminuyendo la detección en un número de ensayos mínimo correspondiente al 95%. No se pudo determinar un SDL para varios plaguicidas a pesar de ser detectados correctamente en las otras matrices diferentes a naranja, pepino, granadilla y uchuva, pues para considerar el SDL se necesitaba la detección al menos en 19 de 20 muestras.
Aplicabilidad
Se analizaron nueve muestras adicionales obtenidas en supermercados, tanto por el método screening descrito previamente, como por una metodología cuantitativa de rutina validada y acreditada bajo la norma ISO 17025. Esta última se basó en el método de extracción sueco para el análisis de residuos de plaguicidas (SweEt) (30). Los resultados de los análisis se muestran en la Tabla 3.
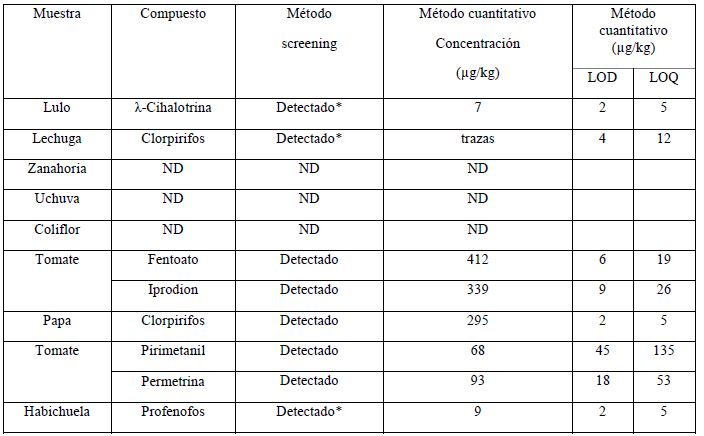
Se observa que en tres muestras (zanahoria, uchuva y coliflor) no se detectó ningún plaguicida por ninguna de las metodologías. En tomate y papa se detectaron los mismos plaguicidas tanto por el método cuantitativo como por el método de screening. Para el caso del lulo se detectó ë-Cihalotrina; en lechuga clorpirifos y en habichuela profenofos tanto por el método cuantitativo como por el método screening, a pesar de que las concentraciones estaban por debajo del SDL. Lo anterior indica la efectividad de la metodología en estudio.
En la Figura 4 se muestran los cromatogramas obtenidos en la detección de clorpirifos en la muestra de lechuga por ambas metodologías. La Figura 4A corresponde al cromatograma de los iones seleccionados del clorpirifos en la detección por el método screening y la Figura 4B a la señal del cromatograma que se obtuvo por el método cuantitativo. En la Figura 4B se superpuso un blanco de matriz (muestra libre de plaguicidas), un nivel de calibración a 30 mg/kg y la señal de clorpirifos en la muestra, lo que demuestra su presencia.
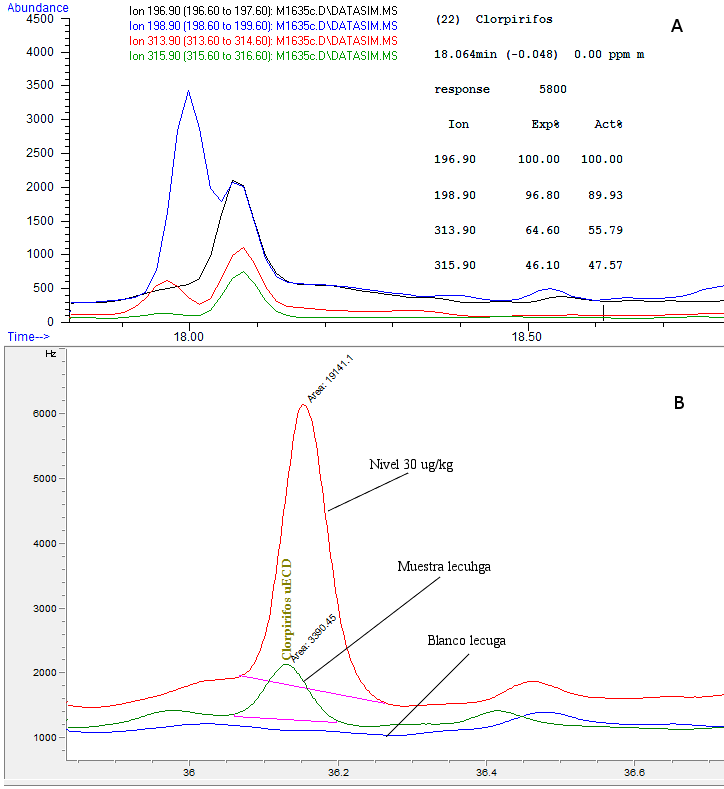
Este plaguicida también fue confirmado por GC/MS en el método cuantitativo. Como se puede observar en la Figura 4A, el método screening permitió la correcta identificación del clorpirifos en lechuga, ya que este cumplió con todos los parámetros de detección establecidos (tiempo de retención, presencia de al menos tres iones, relación intensidades del ion objetivo/iones cualificadores). La Figura 4A también muestra el ion objetivo 196,9 y los iones cualificadores 198,9; 313,9 y 315,9 con las abundancias esperadas y experimentales muy cercanas entre si lo que indica que cumplen con las relaciones de abundancia, confirmando su presencia.
Conclusiones
Se validó una metodología cualitativa para el análisis de residuos de plaguicidas en frutas y hortalizas basada en el método de extracción QuEChERS con un paso de limpieza adicional por cromatografía de permeación por gel (GPC), lo cual permitió reducir la cantidad de componentes de la matriz en el extracto final de algunas matrices, mejorando el desempeño instrumental. La metodología facilita la identificación rápida de 31 plaguicidas al nivel de sus límites máximos de residuos (LMR). El método presenta resultados consistentes respecto a una metodología cuantitativa acreditada en el análisis de frutas y hortalizas.
El método screening permitió la correcta identificación de los residuos en las matrices evaluadas y sus resultados son consistentes con la metodología cuantitativa, por lo tanto, resulta ser una buena alternativa para el análisis de residuos de plaguicidas en este tipo de matrices.
El uso de la cromatografía de gases acoplada a espectrometría de masas con cuadrupolo simple resultó una técnica adecuada y prometedora para el desarrollo de métodos cualitativos rápidos, sin embargo, cabe resaltar la necesidad de una instrumentación más avanzada que permita obtener una mayor selectividad y sensibilidad para determinar mayor número de compuestos.
Agradecimientos
Expresamos nuestro agradecimiento al Departamento de Química de la Universidad Nacional de Colombia, sede Bogotá. Al Instituto Nacional de Metrología (INM) por la financiación para el desarrollo de este trabajo mediante el proyecto 087 llamado “Fortalecimiento de las capacidades de medición nacionales para determinación de contaminantes en alimentos: residuos de plaguicidas en frutas de exportación” y al Organismo Internacional de Energía Atómica (IAEA), Viena, Austria, por la colaboración y soporte obtenido.
Referencias
1. Popp, J.; Peto, K.; Nagy, J. Pesticide productivity and food security. A review. Agron. Sustain. Dev. (2013) 33 (1), 243-255. DOI: https://doi.org/10.1007/s13593-012-0105-x
2. Codex Alimentarius. http://www.fao.org/fao-whocodexalimentarius/standards/pestres/es/ (Consultado el 6 de noviembre de 2016).
3. European Commission Pesticide Data Base. http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=homepage&language=EN. (Consultado el 6 de noviembre de 2016).
4. Regulation of pesticide residues in food US-EPA. https://www.epa.gov/pesticide tolerances (consultado el 6 de noviembre de 2016).
5. Food and Agricultural Materials Inspection Center (FAMIC). https://www.acis.famic.go.jp/eng/chishiki/04.htm (Consultado el 6 de noviembre de 2016).
6. Macbean, E. C. (eds.). A world Compendium The Pesticide Manual, 6th ed. British Crop Production Counsil: Hampshire, UK, 2006; pp 1-557.
7. Fernández-Alba, A. R.; García, J. F. Large-scale multi-residue methods for pesticides and their degradation products in food by advanced LC-MS. TrAC Trends Anal. Chem. 2008, 27 (11), 973–990. DOI: https://doi.org/10.1016/j.trac.2008.09.009.
8. Zamubio, A.; Vanoy, N.; Díaz, C.; Ahumada. D. Desarrolllo y validación de un método multiresiduo para el análisis de plaguicidas en miel por UFLC-MS. Rev.colomb.quim. 2017, 46(2), 24-36. DOI: https://doi.org/10.15446/rev.colomb.quim.v46n2.63014.
9. Guerrero, J.; Velandia, N. Comparación de dos metodologías para la determinación de residuos plagucidas en agua potable. Rev.colomb.quim. 2014, 43(1), 17-24. DOI: https://doi.org/10.15446/rev.colomb.quim.v43n1.50538.
10. Rodrieguez, D.; Diaz, A.; Ahumada, D.; Guerrero, J. Desarrollo y optimización de una metodología multiresiduo por el método simplex para el análisis de plaguicidas en miel de abejas. Rev.colomb.quim. 2013, 42(1), 13-21. DOI: 10.15446/rev.colomb.quim.
11. Bastias, D.; Guerrero, j.; Wyckhuys K. Residuos de plaguicidas en cultivos de pasifloras en regiones de alta producción en Colombia. Rev.colomb.quim. 2013, 42(2), 39-47. DOI: 10.15446/rev.colomb.quim.
12. Portolés, T.; Mol, J. G J.; Sancho, J.; López, V.; Hernández, F. J. Validation of a qualitative screening method for pesticides in fruits and vegetables by gas chromatography quadrupole-time of flight mass spectrometry with atmospheric pressure chemical ionization. Anal. Chim. Acta 2014, 838 ,76-85. DOI: https://doi.org/10.1016/j.aca.2014.06.006.
13. Portolés, T.; Pitarch, E.; López, F. J.; Hernández, F. Development and validation of a rapid and wide-scope qualitative screening method for detection and identification of organic pollutants in natural water and wastewater by gas chromatography time-of-flight mass spectrometry. J. Chromatogr. A 2011, 1228, 303-315. DOI: https://doi.org/10.1016/j.chroma.2010.11.010.
14. Díaz, R.; Ibáñez, M.; Sancho, J. V.; Hernández, F. Target and non-target screening strategies for organic contaminants, residues and illicit substances in food, environmental and human biological samples by UHPLC-QTOF-MS. Anal. Methods 2012, 4, 196-09. DOI: https://doi.org/10.1039/c1ay05385j.
15. Farré, M.; Picó, Y.; Barceló, D. Application of ultra-high pressure liquid chromatography linear ion-trap orbitrap to qualitative and quantitative assessment of pesticide residues. J. Chromatogr. A 2014, 1328, 66-79. DOI: https://doi.org/10.1016/j.chroma.2013.12.082.
16. Rajski, Ł.; Gómez-Ramos, M.; Fernández-Alba, A. R. Large pesticide multiresidue screening method by liquid chromatography-Orbitrap mass spectrometry in full scan mode applied to fruit and vegetables. J. Chromatogr. A 2014, 1360, 119-127. DOI: https://doi.org/10.1016/j.chroma.2014.07.061.
17. López, M.; García, F.; Stead, R.; Roberts, S. L.; McCullagh, D.; Ramesh, M. R. Evaluation and validation of an accurate mass screening method for the analysis of pesticides in fruits and vegetables using liquid chromatography-quadrupole-time of flight-mass spectrometry with automated detection. J. Chromatogr. A 2014, 1373, 40-50. DOI: https://doi.org/10.1016/j.chroma.2014.10.099.
18. Rajski, L.; Gómez-Ramos, M. del M.; Fernández-Alba, A. R. Large pesticide multiresidue screening method by liquid chromatography-Orbitrap mass spectrometry in full scan mode applied to fruit and vegetables. J. Chromatogr. A 2014, 1360, 119–127. DOI: https://doi.org/10.1016/j.chroma.2014.07.061
19. Gómez, M. J.; Gómez-Ramos, M. M.: Agüera, A.; Mezcua, M.; Herrera, S.; Fernández-Alba, A.R. A new gas chromatography/mass spectrometry method for the simultaneous analysis of target and non-target organic contaminants in waters. J. Chromatogr. A 2009, 1216 (18), 4071-4082. DOI: https://doi.org/10.1016/j.chroma.2009.02.085.
20. Mol, H. G. J.; Reynolds, S. L.; Fussell, R. J.; Štajnbaher, D. Guidelines for the validation of qualitative multi-residue methods used to detect pesticides in food. Drug Test. Anal. 2012, 4 (Suppl.1), 10-16. DOI: https://doi.org/10.1002/dta.1364.
21. Norma Técnica Colombiana NTC-ISO/IEC 17025: 2005. Requisitos generales para la competencia de los laboratorios de ensayo y calibración
22. Wylie, P. L; Uchiyama, K. Improved Gas Chromatographic Analysis of Organophosphorus Pesticides with Pulsed Splitless Injection. J AOAC Int. 1996, 79 (2), 571-577.
23. CAC/GL 41-1993. Portion of commodities to which codex maximum residue limits apply and which is analyzed. http://www.fao.org/input/download/standards/43/CXG_041s.pdf. (consultado el 4 de noviembre de 2016).
24. European Standar EN 15662 Foods of plant origin — Determination of pesticide residues using GC-MS and / or LC-MS / MS following acetonitrile extraction / partitioning and cleanup by dispersive SPE — QuEChERS-method. European Committee for Stantarization, 2008; pp 1-83.
25. SANTE/EU. Method validation and quality control procedures for pesticide residues analysis in food and feed, 11945, 2015.
26. European Commission, Deecision 202/657/EC, implementing Council Directive 96/ 23/ EC concerning the performace of analytical methods and the interpretation of results.
27. Lehotay, S. J.; Maštovská, K.; Lightfield, A. R. Use of Buffering and Other Means to Improve Results of Problematic Pesticides in a Fast and Easy Method for Residue Analysis of Fruits and Vegetables. J. AOAC Int. 2005, 88 (2), 615–629.
28. Mol, H. G. J.; Tienstra, M.; Zomer, P. Evaluation of gas chromatography - electron ionization - full scan high resolution Orbitrap mass spectrometry for pesticide residue analysis. Anal. Chim. Acta 2016, 935, 161–172. DOI: https://doi.org/10.1016/j.aca.2016.06.017.
29. Gómez-Ramos, M. M.; Ferrer, C.; Malato, O.; Agüera, A.; Fernández-Alba, A. R. Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables: screening and quantitative studies. J. Chromatogr. A 2013, 1287, 24–37. DOI: https://doi.org/10.1016/j.chroma.2013.02.065.
30. Ross, A. H.; Van Munsteren, A. J.; Nab, F. M.; Tuinstra, L. G. M. Th. Universal extraction/clean-up procedure for screening of pesticides by extraction with ethyl acetate and size exclusion chromatography. Anal. Chim. Acta. 1987, 196, 95-102. DOI: 10.1016/S0003-2670(00)83074-3.
Notas de autor
jaguerrerod@unal.edu.co
Enlace alternativo
https://revistas.unal.edu.co/index.php/rcolquim/article/view/62240 (html)