


Publicado
EFECTO DEL FILTRADO DE SECUENCIAS EN EL ENSAMBLADO DEL GENOMA DE Bacillus altitudinis AISLADO DE Ilex paraguariensis
Effect of sequence filtering on the assembly of the Bacillus altitudinis genome isolated from Ilex paraguariensis
DOI:
https://doi.org/10.15446/abc.v26n2.86406Palabras clave:
análisis de secuencias, biología computacional, control de calidad, genomas bacterianos (es)bacterial genome, computational biology, quality control, sequence analysis (en)
Descargas
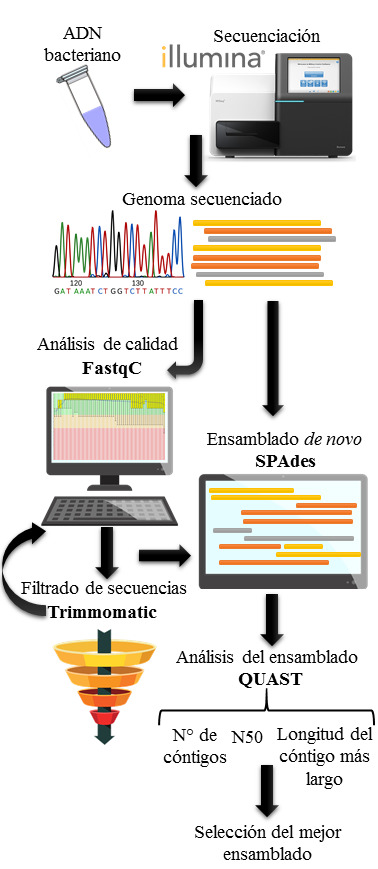
Sin importar el tipo de tecnología aplicada para la secuenciación de un genoma, el filtrado de secuencias es un paso esencial, en el cual aquellas lecturas de baja calidad o parte de estas son eliminadas. En un ensamblado la construcción de un genoma se realiza a partir de la unión de lecturas cortas en cóntigos. Algunos ensambladores miden la relación que existe entre secuencias de una longitud fija (k-mer) que puede verse afectada por la presencia de secuencias de baja calidad. Un enfoque común para evaluar los ensamblados se basa en el análisis del número de cóntigos, la longitud del cóntigo más largo y el valor de N50, definido como la longitud del cóntigo que representa el 50 % de la longitud del conjunto. En este contexto, el presente estudio tuvo como objetivo evaluar el efecto del uso de lecturas crudas y filtradas en los valores de los parámetros de calidad obtenidos en el ensamblado del genoma de la cepa de Bacillus altitudinis19RS3 aislada de Ilex paraguariensis. Se realizó el análisis de calidad de ambos archivos de partida con el softwareFastqC y se filtraron las lecturas con el softwareTrimmomatic. Para el ensamblado se utilizó el softwareSPAdes y para su evaluación la herramienta QUAST. El mejor ensamblado para B. altitudinis19RS3 se obtuvo a partir de las lecturasfiltradas con el valor dek-mer 79, que generó 16 cóntigos mayores a 500 pb con un N50 de 931 914 pb y el cóntigo más largo de 966 271 pb.
Regardless of the type of technology applied to genome sequencing, sequence filtering is an essential step, where those low-quality readings or part of them are eliminated. In an assembly, the construction of a genome is carried out from the union of short readings in contigs. Some assemblers measure the relationship between sequences of a fixed length (k-mer) that can be affected by the presence of low-quality sequences. A common approach to evaluating assemblies is based on the analysis of the number of contigs, the length of the longest contig, and the value of N50 defined as the length of the contig representing 50 % of the length of the assembly. In this context, the present study aimed to evaluate the effect of the use of raw and filtered reads on the values of the quality parameters obtained in the assembly of the genome of the Bacillus altitudinis 19RS3 strain isolated from Ilex paraguariensis. The quality analysis of both starting files was performed with the FastqC software and the readings were filtered with the Trimmomatic software. The SPAdes software was used for the assembly and the QUAST tool for its evaluation. The best assembly for B. altitudinis19RS3 was obtained from the filtered readings with the value of k-mer 79, which generated 16 contigs greater than 500 bp with an N50 of 931 914 bp and the longest contig of 966 271 bp.
Referencias
Aguilar-Bultet L, Falquet L. Secuenciación y ensamblaje de novo de genomas bacterianos: una alternativa para el estudio de nuevos patógenos. Rev Salud Anim. 2015;37(2):125-132.
Andrews S. FastQC a quality control tool for high throughput sequence data. Babraham bioinformatics [monografía en Internet] 2010. Disponible en: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Citado: 30 abr 2020.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455-77. Doi: https://doi.org/10.1089/cmb.2012.0021
Bishop OT. Bioinformatics and data analysis in microbiology. Grahamstown, Sudafrica: Caister Academic Press; 2014. 264 p.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114-2120. Doi: https://doi.org/10.1093/bioinformatics/btu170
Cariaga Martinez AE, Zapata PD. Protocolos de extracción de ADN. El laboratorio de biología molecular. Edición ampliada. Buenos Aires, Argentina: Editorial universitaria; 2007. p. 23-39.
Chen C, Khaleel SS, Huang H, Wu CH. Software for pre-processing Illumina next generation sequencing short read sequences. Source Code Biol Med. 2014;9:8. Doi: https://doi.org/10.1186/1751-0473-9-8
Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. Doi: https://doi.org/10.1186/1471-2105-11-485
Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PloS one. 2013;8(12):e85024. Doi: https://doi.org/10.1371/journal.pone.0085024
Gladman S. De novo Genome Assembly for Illumina Data. Melbourne Bioinformatics [monografía en Internet]. 2019. Disponible en: https://www.melbournebioinformatics.org.au/tutorials/tutorials/assembly/assembly-protocol/#protocol. Citado: 30 abr 2020.
Góngora-Castillo E, Buell CR. Bioinformatics challenges in de novo transcriptome assembly using short read sequences in the absence of a reference genome sequence. Nat Prod Rep. 2013;30(4):490-500. Doi: https://doi.org/10.1039/c3np20099j
Gurevich A, Vladislav S, Nikolay V, Glenn T. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072-1075. Doi: https://doi.org/10.1093/bioinformatics/btt086
Laczeski ME, Onetto AL, Cortese IJ, Mallozzi GY, Castrillo ML, Bich GA, et al. Isolation and selection of endophytic spore-forming bacteria with plant growth promoting properties isolated from Ilex paraguariensis St. Hil. (Yerba mate). An Acad Bras Cienc. 2020; 92. Doi: https://doi.org/10.1590/0001-3765202020181381
Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, et al. Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2016;19(2):286-302. Doi: https://doi.org/10.1093/bib/bbw114
Martin M. Cutadapt removes adapter sequences from high throughput sequencing reads. EMBnet J. 2011;17(19:10-12. Doi: https://doi.org/10.14806/ej.17.1.200
Medvedev P, Pham S, Chaisson M, Tesler G, Pevzner P. Paired de bruijn graphs: a novel approach for incorporating mate pair information into genome assemblers. J Comput Biol. 2011;18(11): 1625-34. Doi: https://doi.org/10.1089/cmb.2011.0151
Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. Doi: https://doi.org/10.1371/journal.pone.0030619
Rana SB, Zadlock FJ, Zhang Z, Murphy WR, Bentivegna CS. Comparison of De Novo Transcriptome Assemblers and k-mer strategies using the killifish, Fundulus heteroclitus. PLoS One. 2016;11(4):e0153104. Doi: https://doi.org/10.1371/journal.pone.0153104
Rodríguez Hernáez JI. Ensamblaje y caracterización genómica de una bacteria celulolítica aislada del rumen bovino (Tesis de Licenciatura en Biotecnología). Buenos Aires: Facultad de Ingeniería y Ciencias Exactas, Universidad Argentina de la Empresa; 2017. 138 p.
Sambrook J, Rusell DW. Molecular cloning: a laboratory manual. Nueva York, USA: Cold spring harbor laboratory press; 2001. 1546 p.
Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One. 2011a;6:e17288. Doi: https://doi.org/10.1371/journal.pone.0017288
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011b;27(6):863–864. Doi: https://doi.org/10.1093/bioinformatics/btr026
Smeds L, Künstner A. ConDeTri-A Content dependent read trimmer for Illumina data. PLoS One. 2011;6:e26314. Doi: https://doi.org/10.1371/journal.pone.0026314
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008; 18:821-829. Doi: https://doi.org/10.1101/gr.074492.107
Cómo citar
APA
ACM
ACS
ABNT
Chicago
Harvard
IEEE
MLA
Turabian
Vancouver
Descargar cita
Licencia
Derechos de autor 2021 Acta Biológica Colombiana

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-CompartirIgual 4.0.
1. La aceptación de manuscritos por parte de la revista implicará, además de su edición electrónica de acceso abierto bajo licencia Attribution-NonCommercial-ShareAlike 4.0 (CC BY NC SA), la inclusión y difusión del texto completo a través del repositorio institucional de la Universidad Nacional de Colombia y en todas aquellas bases de datos especializadas que el editor considere adecuadas para su indización con miras a incrementar la visibilidad de la revista.
2. Acta Biológica Colombiana permite a los autores archivar, descargar y compartir, la versión final publicada, así como las versiones pre-print y post-print incluyendo un encabezado con la referencia bibliográfica del articulo publicado.
3. Los autores/as podrán adoptar otros acuerdos de licencia no exclusiva de distribución de la versión de la obra publicada (p. ej.: depositarla en un archivo telemático institucional o publicarla en un volumen monográfico) siempre que se indique la publicación inicial en esta revista.
4. Se permite y recomienda a los autores/as difundir su obra a través de Internet (p. ej.: en archivos institucionales, en su página web o en redes sociales cientificas como Academia, Researchgate; Mendelay) lo cual puede producir intercambios interesantes y aumentar las citas de la obra publicada. (Véase El efecto del acceso abierto).












