



Publicado
Predicción de la estructura secundaria de proteínas usando Máquinas de soporte Vectorial
Palabras clave:
coding methodology, support vector machines, prediction of protein secondary structure, máquinas de soporte vectorial, metodología de codificación, predicción de la estructura secundaria de proteínas (es)Abstract : Among the computational methods used for predicting secondary structure proteins highlights the use of support vector machines. This research shows the predicted secondary structure of protein from its primary amino acid sequence using Support Vector Machines. As inputs, in the proposed methodology, features are used from different structural motifs or text strings associated with the primary structure which represents the secondary structure, such as R-group and the probability that the amino acid at position adopts a central particular secondary structure. For feature extraction method is used coding of sequences in which each symbol in the primary structure is associated with each symbol in the secondary structure. The use of this encoding method reduces the dimensionality of the data of thousands of characteristics only 220 of these. The results obtained are comparable to those reported in the literature, taking about 70% accuracy. Furthermore, it is possible to reduce computational cost in the construction of classifiers because this work models the problem of multi classification as a group of binary classifiers.
Key words: coding methodology; support vector machines; prediction of protein secondary structure.
Resumen: Entre los métodos computacionales utilizados para la predicción de la estructura secundaria de proteínas, se destaca el uso de máquinas de soporte vectorial. Este trabajo de investigación presenta la predicción de la estructura secundaria de proteínas desde su secuencia primaria de aminoácidos usando Máquinas de Soporte Vectorial. Como entradas, en la metodología propuesta, se utilizan características de los diferentes motivos estructurales o cadenas de texto asociadas a la estructura primaria que representa la estructura secundaria, tales como el R-grupo y la probabilidad de que el aminoácido en la posición central adopte una determinada estructura secundaria. Para la extracción de características se utiliza un método de codificación de secuencias en el que cada símbolo en la estructura primaria se relaciona con cada símbolo en la estructura secundaria. El uso de este método de codificación permite reducir la dimensionalidad de los datos de miles de características a sólo 220 de estas. Los resultados obtenidos son comparables a los registrados en la literatura, teniendo cerca de un 70% de precisión. Además, se logra reducir los costos computacionales en la construcción de los clasificadores debido a que este trabajo modela el problema de multi-clasificación como un grupo de clasificadores binarios.
Palabras clave: máquinas de soporte vectorial; metodología de codificación; predicción de la estructura secundaria de proteínas.
ARTÍCULO CORTO
Predicción de la estructura secundaria de proteínas usando Máquinas de soporte Vectorial
Protein secondary structure prediction using support vector machines
D. J. Delgado1,2, H. Arguello1, R. Torres2.
1 Grupo de Investigación en Ingeniería Biomédica GIIB, Universidad Industrial de Santander, Colombia.
2 Grupo de Investigación en Bioquímica y Microbiología GIBIM, Universidad Industrial de Santander, Colombia
Recibido: mayo 18 de 2011 Aprobado: junio 29 de 2012
Resumen
Entre los métodos computacionales utilizados para la predicción de la estructura secundaria de proteínas, se destaca el uso de máquinas de soporte vectorial. Este trabajo de investigación presenta la predicción de la estructura secundaria de proteínas desde su secuencia primaria de aminoácidos usando Máquinas de Soporte Vectorial. Como entradas, en la metodología propuesta, se utilizan características de los diferentes motivos estructurales o cadenas de texto asociadas a la estructura primaria que representa la estructura secundaria, tales como el R-grupo y la probabilidad de que el aminoácido en la posición central adopte una determinada estructura secundaria. Para la extracción de características se utiliza un método de codificación de secuencias en el que cada símbolo en la estructura primaria se relaciona con cada símbolo en la estructura secundaria. El uso de este método de codificación permite reducir la dimensionalidad de los datos de miles de características a sólo 220 de estas. Los resultados obtenidos son comparables a los registrados en la literatura, teniendo cerca de un 70% de precisión. Además, se logra reducir los costos computacionales en la construcción de los clasificadores debido a que este trabajo modela el problema de multi-clasificación como un grupo de clasificadores binarios.
Palabras clave: máquinas de soporte vectorial, metodología de codificación, predicción de la estructura secundaria de proteínas.
Abstract
Among the computational methods used for predicting secondary structure proteins highlights the use of support vector machines. This research shows the predicted secondary structure of protein from its primary amino acid sequence using Support Vector Machines. As inputs, in the proposed methodology, features are used from different structural motifs or text strings associated with the primary structure which represents the secondary structure, such as R-group and the probability that the amino acid at position adopts a central particular secondary structure. For feature extraction method is used coding of sequences in which each symbol in the primary structure is associated with each symbol in the secondary structure. The use of this encoding method reduces the dimensionality of the data of thousands of characteristics only 220 of these. The results obtained are comparable to those reported in the literature, taking about 70% accuracy. Furthermore, it is possible to reduce computational cost in the construction of classifiers because this work models the problem of multi classification as a group of binary classifiers.
Key words: coding methodology, support vector machines, prediction of protein secondary structure.
Introducción
Las proteínas son macromoléculas poliméricas constituidas por cadenas lineales de 20 diferentes aminoácidos (aa), a estas cadenas se les denomina estructuras primarias, estas cadenas de aa generan tres grandes grupos estructurales al interior de las proteínas: las hélices o estructuras α (H), las láminas o estructuras β (E) y las conformaciones coil (C), ver figura 1. A estas conformaciones se les denomina estructuras secundarias. En este trabajo se propone una metodología de predicción para la estructura secundaria de proteínas que reduzca el número de características medidas para la creación de máquinas de aprendizaje que infieran el contenido estructural de una proteína.

Diversos han sido los métodos computacionales utilizados para predecir la estructura secundaria de una proteína desde su secuencia primaria de aminoácidos; estos métodos incluyen: aquellos basados en la composición de los aminoácidos (Chou, 1980; Nakashima et al, 1985; Muskal and Kim, 1992), redes neuronales (Rost and Sander, 1993b), modelos ocultos Markov (HMM) (Hubbard and Park, 1995) y máquinas de soporte vectorial (MSV).
Hoy en día las MSV están siendo usadas para la solución de problemas en bioinformática, más que otras herramientas. La predicción de la estructura secundaria de proteínas no es la excepción (Hua and Sun, 2001). Predecir la estructura secundaria de una proteína puede analizarse como un típico problema de reconocimiento o clasificación de patrones, en el cual cada aa en la estructura primaria, debe ser clasificado en uno de los tres diferentes grupos estructurales: hélices-α, láminas-β o coil.
Este problema de clasificación se puede abordar mediante la clasificación de los patrones que generan las representaciones textuales de la estructura primaria y secundaria, ver figura 2. Dichas representaciones son generadas por dos alfabetos, el alfabeto que representa la estructura primaria el cual contiene los símbolos que representan a los 20 aminoácidos de los cuales se componen la mayor parte de las proteínas, este alfabeto se denominará ∑ = {A,R,N,D,C,E,Q,G,H,I,L,M,F,P,S,T,W,Y,V}.

El segundo alfabeto, el cual representa simbólicamente los diferentes motivos estructurales que pueden tener los diferentes aminoácidos se denominará Γ = {E,H,C} . Donde representa a las láminas-β, a las hélices-α y a las estructuras coil. Con estos conjuntos de símbolos se puede representar textualmente tanto a la estructura primaria como la estructura secundaria.
Metodología
Las Máquinas de Soporte Vectorial
Para abordar el problema de clasificación de patrones asociado a la predicción de la estructura secundaria de una proteína se necesita de una herramienta matemática que permita clasificar las características extraídas de las cadenas de caracteres que representan las estructuras primarias y que se asocian con su respectiva estructura secundaria. En este trabajo la herramienta usada fueron las máquinas de soporte vectorial (MSV) las cuales son un método efectivo en el área de reconocimiento de patrones en general. Una tarea de clasificación o reconocimiento de patrones generalmente necesita de un conjunto de datos para entrenamiento y otro para realizar las pruebas.
Cada instancia en el conjunto de entrenamiento tiene un valor objetivo (etiqueta de clase) y varios atributos (características). La meta de una MSV es generar un modelo que sea capaz de predecir correctamente los valores objetivo de alguna instancia perteneciente al grupo de pruebas sin conocer cómo está etiquetado, para luego poder extrapolar dicho modelo a cualquier individuo perteneciente al universo del cual se tomaron los ejemplos de entrenamiento.
Para generar un modelo de clasificación se parte de un conjunto de entrenamiento constituido por parejas (xi, yi) i = 1,2,...,/,, donde xi ε Rn e y ε {–1, + 1}/ y / es el número de ejemplos de entrenamiento. Una MSV (Vapnik, 1995; Cortes and Vapnik, 1995) requiere de la solución del siguiente problema de optimización, ver ecuación 1.

Donde WTΦ(xi) + b representa el hiperplano de separación, C controla el equilibrio entre la complejidad de la máquina y el número de puntos no separables por un hiperplano, ξi mide la desviación de un punto xi del punto de separación WTΦ(xi) + b . Los vectores xi son mapeados a un espacio dimensional mayor por la función Φ . Las MSV buscan un hiperplano que realice una separación lineal que tenga un margen de separación máximo entre los grupos a clasificar. C>0 permite el balance entre maximizar el margen y minimizar el error. Además, K(Xi, Xj) ≡ φ(Xi)T φ(Xj) es llamada la función kernel.
La base de datos
Para elaborar el algoritmo de predicción de la estructura secundaria de proteínas es necesario contar con una buena colección de secuencias de péptidos (cadenas de texto que representan la estructura primaria), los cuales se deben usar para poder enseñarle a una máquina de aprendizaje las características que deben tener las diferentes combinaciones de segmentos de proteínas que pueden formar los diferentes motivos estructurales (cadena de texto asociada a la estructura primaria que representa la estructura secundaria). Así como también se deben usar otros péptidos para probar el correcto aprendizaje de las máquinas elaboradas. Para este trabajo se utilizaron dos conjuntos de secuencias de proteínas, uno para poder proporcionar conocimiento y otro para evaluar el conocimiento adquirido. Estos dos conjuntos son los denominados CB513 y el RS126 donde RS216 ? CB513 .
La base de datos CB513 (Cuff and Barton, 1999) consta de 513 secuencias de proteínas en donde todas ellas tienen una longitud mayor a 30 residuos. Esta base de datos fue usada en este trabajo como el conjunto de entrenamiento para las máquinas de aprendizaje desarrolladas excluyendo las 126 secuencias incluidas en ella pertenecientes a la . Mientras que de las RS126 (Rost and Sander, 1993a) se seleccionaron 126 secuencias de proteínas con una homología menor del 25%. Estas bases de datos continúan siendo hoy en día referentes de comparación para el rendimiento de los diferentes algoritmos, pues contienen información no redundante del espacio de proteínas (Sui et al., 2011; Qu et al., 2011; Chatterjee et al., 2011), por lo que se usaron en este trabajo para validar los modelos que se construyeron.
Codificación de las secuencias
Con las dos bases de datos se obtienen los individuos que permitirán tanto implementar como verificar las máquinas de aprendizaje sin embargo, la información presente en estas bases de datos (secuencias de estructuras primarias y secundarias) no se pueden usar directamente y hay que transformar dicha información. Para poder extraer información proveniente de una secuencia de aminoácidos, es necesario convertir dicha cadena en información numérica, vectores que describan el contenido de una secuencia, de un segmento de secuencia o incluso de un aminoácido en particular. Existen diversas formas de codificar la información presente en la estructura primaria de una proteína como diversos son también los problemas en los que se aplican estas codificaciones de la estructura primaria.
Para este trabajo se implementó una metodología para codificar las secuencias que permitiera generar vectores codificados de una dimensionalidad baja y que a su vez trataran de reducir la correlación de los vectores codificados para las diferentes clases existentes. Para ello se emplearon algunos conceptos expuestos por Yang and Wang 2003 y Ruan et al., 2005.
Se necesita relacionar cada símbolo en la estructura primaria con cada símbolo en la estructura secundaria sin embargo, también se debe tener en cuenta a sus vecinos. Para poder extraer dicha información se realiza un ventaneo sobre la secuencia, el resultado de dicho ventaneo es una colección de N-gramas pertenecientes a una misma estructura primaria.
El N-grama
Para obtener información de cada motivo estructural presente en la secuencia de una proteína es necesario recorrer de f1orma adecuada dicha cadena. En Yang and Wang 2003 se muestra una metodología denominada N-grama, la cual se emplea en este trabajo para extraer los segmentos de secuencias pertenecientes a la estructura primaria. Estos segmentos deben ser posteriormente codificados en vectores de características. Para extraer estos segmentos de secuencia se debe tener en cuenta lo siguiente:
O = {O1, O2, …, On} es la estructura primaria de una proteína la cual está compuesta por una cadena de caracteres Oi ε ∑ y n es la longitud de la secuencia.
S = {S1, S2, ..., Sn} es la estructura secundaria la cual está compuesta por otra cadena de caracteres S1 ε Γ de la misma longitud que O.
CS = {(csi,1, csf,1),..., (csi,w, csf,w)} es el conjunto de parejas (csi, csf) que denotan los puntos de inicio (i) y fin (f) de cada una de las subsecuencias de aminoácidos que tienen asociado un mismo símbolo S1 ε S al interior de una misma proteína siendo el número de sub segmentos en ésta. Ver figura 3.

A partir de la cadena O y la cadena S se extraen las posiciones de inicio y fin de cada uno de los segmentos de estructura que pertenecen a un mismo motivo estructural. Cada uno de estos segmentos es una secuencia perteneciente a un motivo estructural al cual se desea codificar.
El N-grama hace referencia a segmentos de N caracteres consecutivos Oi ε ∑ donde el caracter en el centro de esta subcadena es aquel al cual se desea codificar. La forma como se deben extraer dichos N-gramas de las diferentes subsecuencias se puede ver en el algoritmo 1.
Es evidente que aquellos segmentos que se encuentran al inicio y al final de la secuencia corresponden a posiciones fuera del rango de las estructuras primaria y secundaria, estas posiciones en el N-grama deben ser reemplazadas por algún símbolo que permita su posterior codificación.
Codificación de las subsecuencias
Los segmentos de aminoácidos que se obtienen (los N-gramas) deben ser convertidos en vectores de características que permitan plantear un algoritmo de clasificación. Las metodologías empleadas en este trabajo plantean la codificación de las secuencias con base en el VCM y las propiedades de grupo de los aminoácidos, las cuales permitirán descorrelacionar la información que se obtiene. Para convertir en vectores de características los N-gramas extraídos de una secuencia, el procedimiento a seguir es el siguiente: Primero se halla el VCM modificado (VCMM) para un N-grama dado, ver procedimiento para calcular el VCMM en el algoritmo 2, hay que tener en cuenta que se deben hacer ciertas modificaciones sobre el cálculo de dicho vector, dichas modificaciones radican en el cambio del alfabeto sobre el que se realizan los cálculos, el nuevo alfabeto debe contemplar las posiciones nulas del principio y fin de la secuencia en la extracción de los N-gramas (Ruan et al., 2005; Ganapathiraju et al., 2004; Yang and Wang, 2003).

El algoritmo 2, muestra el cálculo del VCMM. Para este trabajo se utilizaron los vectores de orden cero y uno Vcmm=(Xi0, Xi1,), con los cuales se realiza una primera etapa de la codificación de un N-grama. Dado un perteneciente a un N-grama el cual representa un segmento de secuencia en una proteína, se desea dar importancia al caracter central en el N-grama, para ello se le incorporará información estadística perteneciente a dicho caracter. También se desea descorrelacionar los N-gramas de acuerdo a dicho caracter para lo cual se emplearán las propiedades físicoquímicas de los diferentes aa. Este enfoque de codificación considera las probabilidades de que cada caracter en Σ pueda adoptar un determinado tipo estructural Γ dados los diferentes grupos biológicos a los que puede pertenecer cada aminoácido (Nelson and Cox, 2000), ver tabla 1.

La información estadística que se puede agregar a la codificación es la probabilidad de que un aminoácido pueda adoptar una estructura Γ dada una de las clasificaciones físicoquímicas en las que se pueden clasificar cada aminoácido. Estas clasificaciones se codificarán y se les llama los R-grupos, los cuales también forman parte de la codificación, ver tabla 1. Estas probabilidades se pueden encontrar de la siguiente manera: dado un conjunto de entrenamiento Δ y un conjunto de grupos C = {C1, C2, C3, C4, C5} en los que se puedan clasificar los aminoácidos, la probabilidad de que un residuo aai en Ci para j = 1, 2,...,5 sea una hélice (E), una lámina (H) o una conformación coil (C) es, ver ecuación 2:
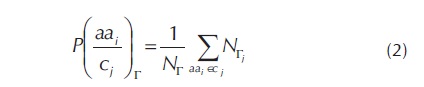
Donde P[aai/cj]Γ para i = 1, 2, ...,20 es la probabilidad de que el residuo aai dado un grupo Cj esté en Γ, es decir la probabilidad de que el residuo aai sea una hélice P[Oc/ci]H, una lámina P[Oc/cj]E o Coil P[Oc/cj]c en un conjunto de entrenamiento Δ. NΓ es el número total de residuos de cada una de las diferentes conformaciones H, E y C que hay en Δ, y NΓ es el número en el que el residuo aai que pertenece a el grupo Ci adopta una conformación Γ.
En este trabajo se usa el producto de Kronecker (Zwillinger 1996) entre las probabilidades encontradas, los vectores de codificación que obtienen de los R-grupos y los VCMM que se calculan a partir de los N-gramas para descorrelacionar los vectores de características (Yang and Wang, 2003). Obteniendo de esta forma la codificación de los segmentos de los aminoácidos para ser usados más adelante. La codificación de las secuencias se puede ver en la ecuación 3.

En donde P[aai/cj]Γ es la probabilidad de que el aminoácido en la posición central de un N-grama adopte una determinada estructura secundaria dado uno de los diferentes Ci grupos en los que se pueden agrupar los diferentes residuos. Ci es la codificación del aminoácido en la posición central del N-grama (el R-grupo) y Vcmm es el VCMM que se calcula del N-grama, el operador (X) representa el producto elemento a elemento entre dos vectores, y el operador representa el producto de Kronecker.
Una vez codificados los N-gramas, lo que se busca es encontrar funciones Fs, ver ecuación 4, que permitan asociar vectores Γ con una de las diferentes estructuras Γ ε{C,E,H} .

Planteamiento del problema
Sea VCS el conjunto de posibles vectores codificados pertenecientes a N-gramas extraídos de secuencias de proteínas empleadas como ejemplos de entrenamiento. Sea Γ el conjunto finito de clases en las que se pueden clasificar los ejemplos VCS y k el tamaño de Γ (k=3, C, E, H). Formalmente el algoritmo de aprendizaje (para este trabajo MSV) toma un conjunto de ejemplos de entrenamiento ((v1, y1), (v2, y2),...,(vm, ym)) como entradas, donde y1ε Γ son las etiquetas asignadas a los ejemplos de entrenamiento v1 ε VCS. El objetivo del algoritmo de aprendizaje es generar una hipótesis f:VxΓ → R donde f pertenece al espacio de hipótesis F.
El algoritmo de clasificación a utilizar son las MSV, las cuales son clasificadores binarios y el problema de clasificación que se tiene cuenta con más de dos clases, para problemas binarios ( clases) los ejemplos son etiquetados como -1 y +1, por conveniencia. Lo que se busca es generar una hipótesis f : V ? {-1, + 1} . Por tanto se debe adecuar un problema de multi-clasificación en términos de problemas de clasificación binaria.
Descripción de la solución
Un problema de multi-clasificación se puede reducir a múltiples problemas de clasificación binarios los cuales se pueden resolver separadamente. Existen diversas formas de reducir un problema de multi-clasificación en problemas de clasificación binaria (Trevor and Tibshirani, 1998; Dietterich and Bakiri, 1994), uno de ellos indica que a cada clase kεΓ se puede asociar con una fila de una matriz de codificación M ε {-1,0,1}k×l la cual relaciona los diferentes clasificadores binarios fs que se pueden conformar mediante combinaciones de las clases Γ en las cuales se desea clasificar, en esta matriz se muestran las respuestas que se esperan de cada clasificador binario cuando los datos provienen de una clase en particular, ver tabla 2. Donde I representa el número de clasificadores binarios fs que se crearon empleando un algoritmo de aprendizaje, para S = 1,2,...,l,, además l también representa el número de clasificadores en los que se puede descomponer el problema de multiclasificación. Los clasificadores binarios S se pueden desarrollar teniendo en cuenta el enfoque de emparejamiento total (Allwein et al., 2000) de las k clases. Para este problema en particular se tiene I=(k/2) clasificadores (f1 = E|H, f2 = E|C, f3 = C|H). Los clasificadores fs son entrenados para cada columna de la matriz M, es decir cada columna de la matriz de codificación contempla un problema de clasificación binaria donde las etiquetas (vi, M(yi.s)) indican cuales son los ejemplos de entrenamiento para cada clasificador fs . Los datos donde (vi, M(yi.s)) = 0 no se contemplan para el entrenamiento, pues son aquellos datos que no corresponden al clasificador binario en cuestión.

Para el entrenamiento de los diferentes clasificadores binarios, los cuales se muestran en las columnas de la matriz de codificación M, se debe entrenar una MSV las cuales tienen dos parámetros que se deben ajustar, C y γ, donde γ es el parámetro libre de la función Kernel que se usó, en este caso la función de base radial (RBF), verecuación 5.

Se tomó como función Kernel la RBF debido a que en diversos trabajos esta función es la que mejores resultados ha ofrecido (Shoyaib et al., 2007; Chen et al., 2006; Hua and Sun, 2001; Cai et al., 2001). Como se tienen dos parámetros libres, el problema es encontrar qué valores deben asumir estos dos parámetros para encontrar el mejor clasificador. El objetivo es identificar (C,&gamma) tales que el clasificador sea capaz de predecir adecuadamente los datos de prueba, es decir aquellos que no se utilizan para generar el modelo. Se recomienda una combinación de los diferentes parámetros C y γ, para ello se toma un intervalo de estos dos parámetros C y g donde Cinicial y Cfinal así como γinicial y γfinal denotan los límites entre los cuales se desea probar las MSV, ΔC y Δγ es el paso que se toma para construir los intervalos y m es el número de muestras que se desea tomar.

Lo que se busca es encontrar la combinación (Ci,?j) que genere la MSV que tenga el mejor rendimiento Qi,j (vertabla 3). Realizado esto con las MSV que se deben entrenar con los datos que proporciona la matriz M, se tendrán los diferentes clasificadores binarios que se van a utilizar.

El problema de multi-clasificación, se necesita que, para un ejemplo Vcs, se pueda saber a qué clase K pertenece. Para ello se utiliza el enfoque denominado códigos de corrección de errores de salida (por sus siglas en inglés ECOC) (Dietterich and Bakiri, 1994). Tomando M(k) como una fila de la matriz de codificación y sea f(Vi) el vector de las predicciones que se obtienen de los clasificadores fs para un vector V.

La forma de encontrar la clase kεΓ de cualquier vector f(V) es encontrando la fila de M que minimice la distancia d(M(k), f(V)) para alguna distancia d . Para medir estas distancias y encontrar las clases a las cuales se le puede asociar un dato V, se puede realizar mediante una función de pérdida L, ver ecuación 9, la cual mide el margen de pérdida cuando un clasificador fs es evaluado con un ejemplo Vi respecto a M(yi, s). La función L se evalúa sobre sobre las diferentes filas de la matriz M .

Se selecciona la clase k que más coincida con las predicciones realizadas por los diferentes clasificadores fs, para ello mediante el uso de la función de pérdida L se calculan las distancias entre el vector f(V) y las filas de la matriz M, con las cuales se quiere buscar a qué clase k pertenece el vector V, esta clase k es la que tenga la mínima de las distancias y ver ecuación 10. En donde y permite inferir con cuál de las filas de la matriz M tiene una mayor similitud el vector de resultados del clasificador f, con lo cual se puede también inferir en qué clase se va a clasificar el vector V . Este enfoque es denominado decodificación basada en pérdida (Allwein et al., 2000).
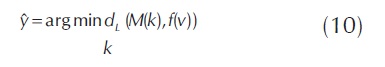
Implantación de la solución
Dados los 3 clasificadores fs y la estrategia para combinarlos con el fin de generar un multiclasificador, lo que se busca es crear una metodología que permita clasificar subsecuencias de caracteres que representan aminoácidos, donde a dichas subsecuencias no se les conoce su estructura secundaria. Los pasos descritos en apartados anteriores, dicen que se deben determinar los N-gramas contenidos al interior de la secuencia que representa a una proteína, (ver algoritmo 2), se debe a partir de dicha subsecuencia y más concretamente del caracter que se encuentra justo en el centro de ésta, clasificar este aminoácido en uno de los grupos mostrados en la tabla 1, de donde se obtiene el R-grupo y, además de este mismo aminoácido, interesa encontrar la probabilidad P[aai/cj]Γque adopte.
Dichas probabilidades en la etapa de entrenamiento son fáciles de calcular debido a que se conoce a qué tipo de estructura pertenece un determinado aminoácido. Sin embargo, para realizar la predicción sólo se cuenta con la estructura primaria de la proteína. Por lo cual se plantea una forma de calcular dichas probabilidades con el siguiente enfoque:
Sea ÔεO un N-grama, sea Oc el caracter que se ubica en la parte central de Ô, lo que se busca es que con base en la información que se pueda extraer de Ô predecir a qué tipo de estructura secundaria Γ pertenece Oc . De acuerdo al enfoque mostrado en este trabajo se debe calcular la probabilidad P[aai/cj]Γ, donde Oc es el caracter central, Oi es la clasificación que se puede realizar sobre los caracteres Σ, ver tabla 1. Sin embargo γi no se conoce. Para ello se supone que Oc puede adoptar cualquiera de las estructuras secundarias γ, se van a calcular P[Oc/ci]E, P[Oc/ci]H y P[Oc/ci]C, para poder hacer uso de los clasificadores fs, ver algoritmo 3, con lo cual se puede inferir qué tipo de estructura secundaria puede tomar Oc .

Resultados
Entrenamiento y pruebas
En la validación de los modelos elaborados para predecir la estructura secundaria de una proteína se utilizó la base de datos denominada RS125, para la cual se codificaron todas sus secuencias y se evaluaron en las MSV entrenadas con la base de datos CB513. Para medir la capacidad de estas máquinas en la interpretación de los patrones que se encuentran en la estructura primaria de las proteínas y así poder comparar su efectividad con los enfoques elaborados en otros trabajos de investigación similares.
Medidas de rendimiento
La medida de rendimiento que se utilizó para evaluar los modelos elaborados para cada algoritmo de clasificación es la que se usa usualmente y se define como.
Donde la función P(Γ) calcula el número de aciertos en las diferentes clases Γ y N es el número de ejemplos para prueba (125). También se evaluaron por separado cada uno de los clasificadores fs para ello se empleó el coeficiente de correlación de Mathews (CCM) (Baldi et al., 2000), la sensibilidad (Sens) y la especificidad (Espc) de cada una de las máquinas creadas, ver ecuaciones 12 y 13 . Donde la sensibilidad y la especificidad son mediciones probabilísticas sobre los clasificadores que se crearon. La sensibilidad mide la proporción de verdaderos positivos (aminoácidos correctamente clasificados), la especificidad mide la proporción de aspectos negativos que han sido identificados correctamente. Para las ecuaciones 12, 13 y 14 se tiene que VP representa a los aminoácidos que son correctamente clasificados en una clase determinada, FN representa a los aminoácidos que sin pertenecer a una clase se clasifican como no pertenecientes a ellas, VN representa aquellos aminoácidos que perteneciendo a una clase son identificados como no miembros y FP son aquellos aminoácidos que siendo no miembros de una clase son identificados como miembros de ella. Adicionalmente el coeficiente de correlación de Mathews proporciona una medida de la calidad de los clasificadores binarios que se crearon, un CCM de 1 indica que se construyó un clasificador binario eficiente y un CCM de 0 indica que el clasificador fue deficiente.
Resultados obtenidos
Luego de montar la infraestructura de los diferentes clasificadores y haberlos configurado como un solo clasificador, se procedió a evaluar su rendimiento de acuerdo a las medidas de rendimiento antes mencionadas. Primero se evaluó cómo fue el comportamiento global y también el de cada una de las clases a clasificar, ver tabla 4.

Se evaluó también el rendimiento de cada uno de los clasificadores fsi para poder tener una noción más detallada del funcionamiento del clasificador general. Ver tabla 5.

Se realizó la comparación del rendimiento global del clasificador con algunos trabajos realizados por otros autores, los cuales emplearon las mismas bases de datos para realizar los procesos de entrenamiento y validación. Vertabla 6.

Discusión de resultados
Los resultados obtenidos por el modelo de clasificación construido dejan entrever el grado de dificultad que existe para dar solución al problema de la predicción de la estructura secundaria de proteínas, empleando cadenas de texto para inferir los diferentes motivos estructurales.
En la tabla 4 se muestra el rendimiento global del clasificador, así como también se muestra el rendimiento que dicho clasificador obtiene con cada uno de los motivos estructurales a clasificar. En esta tabla se evidencia que la clase que mayor dificultad presenta es aquella etiquetada con el caracter C . Si se observa la tabla 5, el cual muestra con más detalle el clasificador. Esto debido a que se muestra cómo es el rendimiento de cada clasificador binario. Se observa en la tabla un comportamiento especial en el clasificador fHC en el cual el CCM y la Espc arrojan valores que sugieren que para los grupos etiquetados con los caracteres H y C existe un desbalanceo de información, lo que lleva a que se presente este comportamiento.
El método de codificación empleado permite generar vectores de características de una dimensionalidad baja en comparación a otros métodos existentes actualmente, alcanzando valores de rendimiento similares a aquellas metodologías que por su gran dimensionalidad en sus vectores de características aseguran la descorrelación entre las diferentes clases pero hacen del proceso de clasificación una tarea más difícil, ver tabla 6.
No es posible realizar una comparación del costo computacional o la cantidad de recursos que se consumen en la clasificación debido a que otros autores no incluyen este tipo de información, pero además de obtener resultados comparables a los obtenidos por otros autores como se menciona anteriormente, el hecho de modelar el problema como un conjunto de clasificadores binarios y utilizar un vector de características de menor dimensionalidad produce reducción del costo computacional.
Los resultados obtenidos muestran un porcentaje menor de rendimiento sin embargo, cabe resaltar que en los conjuntos de validación, la base de datos RS126 está contenida dentro de la base de datos CB513. Los estudios muestran que se realiza el entrenamiento de las diferentes máquinas de aprendizaje empleadas en la literatura con el conjunto de datos CB513 y la validación con el conjunto RS126. Lo cual muestra que las máquinas en cierta forma conocen los datos con los cuales serán evaluadas. En este trabajo se excluyó el conjunto de datos RS126 del conjunto de secuencias CB513, por lo cual aseguramos que los resultados obtenidos son producto de la generalización producida por el entrenamiento de los datos y no por la memorización de estos.
Conclusiones
Los resultados obtenidos con las máquinas de soporte vectorial para la predicción de la estructura secundaria de una proteína ratifican la capacidad de esta herramienta para llevar a cabo minería de datos o predicción, en este caso el éxito de las predicciones estuvo cercano al 65%, lo cual para este tipo de problema se considera un rendimiento aceptable.
La herramienta asegura que es capaz de encontrar los hiperplanos de separación óptimos para dos conjuntos de datos cualesquiera, presentando variaciones en el rendimiento dependiendo de cómo se ajusten los parámetros libres presentes en el modelo sin embargo, el éxito de las MSV radica en gran medida en la manera como se codifiquen dichos datos y en cómo dicha codificación asegure la menor correlación entre las clases presentes.
El uso del método de codificación de las secuencias permitió generar vectores de características de una dimensionalidad baja y la reducción de correlación entre las diferentes clases, lo que en todo problema de clasificación conlleva a una reducción del costo computacional.
El rendimiento global obtenido es comparable con los resultados obtenidos por otros autores, pero se destaca que reduce la dificultad del proceso de clasificación al crear un vector de características de muy baja dimensión, pasando de miles de características a 220 de estas.
Los rendimientos promedio de la mayoría de soluciones en esta área de investigación dejan entrever que es una problemática no resuelta aún y que los resultados obtenidos por un solo modelo de clasificación no son confiables. Por tanto, el uso de este tipo de herramientas es útil cuando se realizan las predicciones con diferentes herramientas y que con base en los resultados obtenidos por todos ellos, se puede tomar una decisión acerca de cuál podría ser el contenido estructural presente en una proteína.
Para mejorar los resultados obtenidos, se propone mejorar la selección de los parámetros libres en las máquinas de aprendizaje, así como la incorporación de información inherente al contexto biológico que pueda incrementar la precisión del método. Se propone además, que en el conjunto de características obtenido a partir de la codificación propuesta, se apliquen métodos como el análisis de componentes principales (PCA) en pro de reducir la información redundante y aún más la información presente en los vectores de codificación y así disminuir los tiempos de cómputo.
Referencias bibliográficas
1 Allwein E.L., Schapire R.E., and Singer Y. 2000. Reducing multiclass to binary: a unifying approach for margin classifiers. Journal of Machine Learning Research. 1:113-141.
2 Baldi P., Brunak S., Chauvin Y., Andersen C.A.F. and Nielsen H. 2000. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 16:412-424.
3 Cai Y.D., Liu X.J., Xu X.B. and Zhou, G.P. 2001. Support vector machines for predicting protein structural class.BMC Bioinformatics. 2:3.
4 Chatterjee P., Basu, S., Kundu, M., Nasipuri, M., and Plewczynski, D. 2011. PSP_MCSVM: brainstorming consensus prediction of protein secondary structures using two-stage multiclass support vector machines.Journal of Molecular Modeling. 17(9):2191-2201.
5 Chen C., Tian Y., Zou X., Cai P., and Mo J. 2006. Prediction of protein secondary structure content using support vector machine. Talanta. 71(5): 2069-2073.
6 Chou P.Y. 1980. Amino acid composition of four classes of proteins. Second Chemical Congress of the North American Continent, Las Vegas, Nevada.
7 Cortes C. and Vapnik V. 1995. Support-vector networks. Machine Learning. 20(3):273-297.
8 Cuff J.A., Barton G.J. 1999. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins: Structure, Function, and Bioinformatics. 34(4): 508-519.
9 Dietterich T.G. and Bakiri G. 1994. Solving multiclass learning problems via error-correcting output codes.Journal of Artificial Intelligence Research. 2(1): 263-286.
10 Ganapathiraju M.K., Klein-Seetharaman J., Balakrishnan N. and Reddy, R. 2004. Characterization of protein secondary structure. IEEE Signal Processing Magazine. 21(3): 78-87.
11 Garnier J., Osguthorpe D.J. and Robson, B. 1978. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. Journal of Molecular Biology. 120(1): 97-120.
12 Hua S. and Sun Z. 2001. A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. Journal of Molecular Biology. 308(2): 397-407.
13 Hubbard T.J., Park J. 1995. Fold recognition and ab initio structure predictions using hidden markov models and beta-strand pair potentials. Proteins. 23(8): 398-402.
14 Muskal S.M. and Kim, S.H. 1992. Predicting protein secondary structure content: A tandem neural network approach. Journal of Molecular Biology. 225(3): 713-727.
15 Nakashima H., Nishikawa K. and Ooi T. 1985. The folding type of a protein is relevant to the amino acid composition. Oxford Journals Life Sciences. The Journal of Biochemistry. 99(1): 153-162.
16 Nelson, D. and Cox, M. 2000. Lehninger principles of biochemestry. W.H. Freeman and company. New York. 1152.
17 Qu W., Yang B., Jiang W. and Wang L. 2011. HYBP_PSSP: a hybrid back propagation method for predicting protein secondary structure. Neural computing and applications. 21(2):337-349.
18 Rost B. and Sander C. 1993a. Improved prediction of protein secondary structure by use of sequence profiles and neural networks. Proceedings of the National Academy of Sciences of the United States of America. Biophysics. 90:7558-7562.
19 Rost B. and Sander C. 1993b. Prediction of protein secondary structure at better than 70% accuracy. Journal of Molecular Biology. 232(2): 584-599.
20 Ruan J., Wang K., Yang J., Kurgan L.A. and Cios K.J. 2005. Highly accurate and consistent method for prediction of helix and strand content from primary protein sequences. Artificial Intelligence in Medicine. 35(1-2): 19-35.
21 Shoyaib M., Baker S., Jabid T., Anwar F. and Khan H. 2007. Protein secondary structure prediction with high accuracy using support vector machine. 10th International Conference on Computer and Information Technology. 1-4.
22 Sui H., Qu W., Yan B., and Wang L. 2011. Improved protein secondary structure prediction using an intelligent HSVM method with a new encoding scheme. International Journal of Advancements in Computing Technology. 3(3):239-250.
23 Trevor H. and Tibshirani. 1998. Classification by pairwise coupling. The Annals of Statistics. 26:451-471.
24 Vapnik, V.N. 1995. The nature of statistical learning theory. Springer-Verlag New York.
25 Yang X. and Wang B. 2003. Weave amino acid sequences for protein secondary structure prediction. Proceedings of the 8th ACM SIGMOD workshop on Research issues in data mining and knowledge discovery. 80-87.
26 Zhang G.Z., Huang D.S., Zhu, Y.P., and Li, Y.X. 2005. Improving protein secondary structure prediction by using the residue conformational classes. Pattern Recognition Letters. 26(15): 2346-2352.
27 Zwillinger, D. 1996. Standard mathematical tables and formulae. University of California. 30th Edition. CRC Press. p 812.
Cómo citar
APA
ACM
ACS
ABNT
Chicago
Harvard
IEEE
MLA
Turabian
Vancouver
Descargar cita
Visitas a la página del resumen del artículo
Descargas
Licencia
Derechos de autor 2012 Revista Colombiana de Biotecnología

Esta obra está bajo una licencia internacional Creative Commons Atribución 4.0.
Esta es una revista de acceso abierto distribuida bajo los términos de la Licencia Creative Commons Atribución 4.0 Internacional (CC BY). Se permite el uso, distribución o reproducción en otros medios, siempre que se citen el autor(es) original y la revista, de conformidad con la práctica académica aceptada. El uso, distribución o reproducción está permitido desde que cumpla con estos términos.
Todo artículo sometido a la Revista debe estar acompañado de la carta de originalidad. DESCARGAR AQUI (español) (inglés).















