



Published
Report of a novel mutation in the SLC26A2 gene foud in a colomian adult patient with diastrophic dysplasia
Reporte de una nueva mutación en el gen SLC26A2 en un paciente adulto colombiano con displasia diastrófica
Keywords:
Colombia, Osteochondrodysplasias, Transcriptional Activation, Mutation (en)Activación Transcripcional, Colombia, Mutación, Osteocondrodisplasias (es)
Downloads
Background. Diastrophic dysplasia is an osteochondrodysplasia belonging to the group of dysplasias caused by mutations in the diastrophic dysplasia sulfate transporter. This sindrome is a micromelic dysplasia with multiple bone deformities of the hands, feet, knees and spine.
Objective. Describe the first report of diastrophic displasia in Colombia
Materials and methods. In this paper a Colombian adult patient with diastrophic dysplasia whose clinical diagnosis was confirmed at the molecular level is reported.
Results. In this first report of diastrophic dysplasia in Colombia we found that the patient was compound heterozygote for the already reported Arg279Trp substitution and an unpublished mutation, a Ser157Thr substitution in the SLC26A2 gene.
Conclusion. Bioinformatic analysis on the latter mutation suggested that it could correspond to a deleterious mutation because it is in a highly conserved domain of the sulfate transporter.
Antecedentes. La displasia diastrófica es una osteocondrodisplasia que pertenece al grupo de las enfermedades genéticas del esqueleto causadas por mutaciones en los transportadores de sulfato. Se presenta como una displasia micromélica con afectación de múltiples huesos y deformidades en manos, pies, rodillas y columna vertebral.
Objetivo. Describir el primer reporte de displasia diastrófica en Colombia.
Materiales y métodos. Se reporta un adulto colombiano con esta displasia, con confirmación clínica, radiológica y molecular.
Resultados. En este primer reporte colombiano, se encontró que la paciente presentó una mutación heterocigota compuesta, de Arg279Trp y Ser157Thr, esta última no reportada previamente, en el gen SLC26A2.
Conclusión. El análisis bioinformático de la mutación nueva sugiere que podría corresponder a una mutación deletérea dado que el dominio afectado en el transportador de sulfatos es altamente conservado.
Report of a novel mutation in the SLC26A2 gene found in a Colombian adult patient with diastrophic dysplasia
Reporte de una nueva mutación en el gen SLC26A2 en un paciente adulto colombiano con displasia diastrófica
Tatiana Pineda¹ • Antonio Rossi² Luisa Bonafè³ • Andrea Superti-Furga³ • Harvy M. Velasco MSc¹
1 Instituto de Genética, Universidad Nacional de Colombia. Bogotá.
2 Department of Molecular Medicine, Section of Biochemistry, University of Pavia. Italy.
3 Centre Hospitalier Universitaire Vaudois. Lausanne, Switzerland.
Correspondencia: hmvelascop@unal.edu.co
Recibido: 24/04/2013 / Aceptado: 19/07/2013
Summary
Background. Diastrophic dysplasia is an osteochondrodysplasia belonging to the group of dysplasias caused by mutations in the diastrophic dysplasia sulfate transporter. This sindrome is a micromelic dysplasia with multiple bone deformities of the hands, feet, knees and spine.
Objective. Describe the first report of diastrophic displasia in Colombia
Materials and methods. In this paper a Colombian adult patient with diastrophic dysplasia whose clinical diagnosis was confirmed at the molecular level is reported.
Results. In this first report of diastrophic dysplasia in Colombia we found that the patient was compound heterozygote for the already reported Arg279Trp substitution and an unpublished mutation, a Ser157Thr substitution in the SLC26A2 gene.
Conclusion. Bioinformatic analysis on the latter mutation suggested that it could correspond to a deleterious mutation because it is in a highly conserved domain of the sulfate transporter.
Key words: Colombia, Osteochondrodysplasias, Transcriptional Activation, Mutation. (MeSH).
Resumen
Antecedentes. La displasia diastrófica es una osteocondrodisplasia que pertenece al grupo de las enfermedades genéticas del esqueleto causadas por mutaciones en los transportadores de sulfato. Se presenta como una displasia micromélica con afectación de múltiples huesos y deformidades en manos, pies, rodillas y columna vertebral.
Objetivo. Describir el primer reporte de displasia diastrófica en Colombia.
Materiales y métodos. Se reporta un adulto colombiano con esta displasia, con confirmación clínica, radiológica y molecular.
Resultados. En este primer reporte colombiano, se encontró que la paciente presentó una mutación heterocigota compuesta, de Arg279Trp y Ser157Thr, esta última no reportada previamente, en el gen SLC26A2.
Conclusión. El análisis bioinformático de la mutación nueva sugiere que podría corresponder a una mutación deletérea dado que el dominio afectado en el transportador de sulfatos es altamente conservado.
Palabras clave: Activación Transcripcional, Colombia, Mutación, Osteocondrodisplasias. (DeCS).
Introduction
Diastrophic dysplasia is an autosomal recessive osteochondrodysplasia described since 1960; at present, it belongs to Group 4 of the classification of genetic skeletal disorders (1). The term "diastrophic" was borrowed from geology by Lamy and Maroteaux (2), where diastrophism refers to the process of deformation of the earth´s crust where by geological structures are formed (3).
This syndrome is a micromelic dysplasia with shortening of the limbs, normal sized head, mild shortening of the trunk, "hitchhiker" thumb that is caused by malformations in the first metacarpal, narrow chest, protruding abdomen, spinal deformities, contractures of large joints, dislocation of the radius, cleft palate (in 1/3 of cases) and external ear deformities. Other features include ulnar deviation of fingers, separation between the first and second toe, clubfoot and hemangiomas on the forehead (4) (5).
In 1994 Hästbacka et al. mapped by linkage analysis the gene causing diastrophic dysplasia, the SLC26A2, also known as DTDST (diastrophic dysplasia sulfate transporter), in the locus 5q32 (6). The gene encodes for a 739 amino acid protein with 12 transmembrane domains that acts as a sulfate carrier not dependent on sodium (7). This protein is a sulfate/chloride exchanger of the cell membrane; functional impairment lead to cellular depletion of sulfate and a consequent failure in the sulfation of cartilage proteoglycans (8).
The clinical spectrum of mutations in this gene is very heterogeneous, ranging from lethal forms as Type 2 atelosteogenesis (AO2) and Type 1B Achondrogenesis (ACG1B) to nonlethal forms such as diastrophic dysplasia (DTD) and recessive multiple epiphyseal dysplasia (rMED), the latter being the mildest form of the disease (9). We present the first case reported in the national literature and perform a phenotypic and genotypic analysis of a 34 years old female patient with normal cognition.
Case report
34 years old patient, the first offspring of nonconsanguineous Colombian parents. The mother reported that she had prenatal care during pregnancy without performing ultrasounds and thus without prenatal diagnosis. The birth was by c-section and no data were obtained from neonatal adaptation. The neonatal evaluation showed bilateral clubfoot and anomalies in the right outer ear. Psychomotor development with head support at 3 months, sitting at 8 months, crawled at 12 months and standing at 26 months; language development was adequate.
During childhood the patient started presenting various clinical problems, mostly related to orthopedic complications, but no hospitalization for respiratory events was required. At 2 years of age, she developed scoliosis, when she was 22 years old she was diagnosed with rupture of both menisci, recurrence of clubfoot which was refractory to surgery. Osteoarthritis of the hip was diagnosed at 31 years of age.
For the management of orthopedic complications she required several surgeries, including the correction of clubfoot 2 times at age 2 and 6, vertebral resection at age 3, right knee arthroscopic at age 26 and 27, right meniscus tear repair and excision of pilonidal cyst at age 21. The menarche occurred at age 14 and currently she has regular menstrual cycles (G0P0A0).
Her parents are not consanguineous; the only skeletal sign comes from the father who has ectrodactyly of left hand (Figure 1).

Physical examination showed a normocephalic patient with normal facial phenotype. The patient´s height was 110 cm, span 92 cm, weight 40 kg, head circumference 57 cm, upper segment of 70 cm and lower segment of 40 cm, with an upper/lower segment ratio of 1.75. Right outer ear dysplasia is evident (Figure 2). In the hands, she has clinodactyly, first and second phalanx with apparent symphalangism and bilateral thumb abduction, showing the so-called "hitchhiker" thumb (Figure 3).


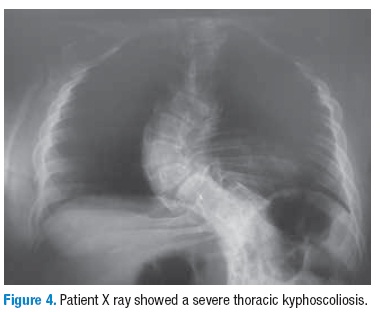
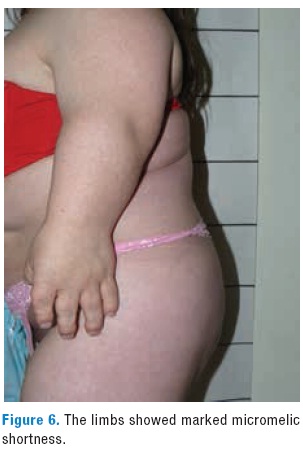
In the back xyphosis and rotoscoliosis is evident in the chest radiography and in the CT (Figures 4-5); in the limbs it is remarkable a severe rhizhomelic and moderate mesomelic and acromelic shortening (Figure 6). The patient also has evidence for mobility restriction of the knees and bilateral clubfoot (Figure 7). There are no neurological deficit and her IQ and social interaction are normal.


Molecular analysis showed that the patient was compound heterozygote for the already reported Arg279Trp substitution and an unpublished mutation, a Ser157Thr substitution in the SLC26A2 gene. In addition, the patient was homozygous for the Thr689Ser polymorphism. The so-called "Finnish allele", a pathological mutation outside the coding sequence, was not detected. The father was heterozygous carrier of the Arg279Trp substitution, while the mother carried the Ser157Thr mutation.
Discussion
Diastrophic dysplasia (OMIM # 222600) is a clinical syndrome classified within the sulfation disorder group in the current "Nosology and Classification of Genetic Skeletal Disorders: 2010 Revision" (1). In Latin America, its prevalence is 0.13 in 10000 newborns according to data from Barbosa et al. taken from ECLAMC (2000-2007) (10), while the highest incidence is in Finland, with 1 case per 30000 newborns. In this paper, we report the first Colombian patient with clinical and molecular diagnosisof diastrophic dysplasia.
Patients with diastrophic dysplasia have an increased mortality in the neonatal period due to respiratory complications resulting from a small chest and a collapsible trachea; in adulthood, the disease affects the bone structures, tendons, ligaments and joint capsules, causing mobility restriction. Frequently, joint contractures and spinal deformities worsen with age, leading to the onset of scoliosis, lumbar hyperlordosis and thoracolumbar kyphosis.
Our case had several typical features of diastrophic dysplasia like calcification of the right ear, hitchhiker thumb and orthopedic complications such as shortening of the limbs, bilateral clubfoot refractory to surgery and severe spinal deformities evidenced by xyphosis and rotoscoliosis. The patient did not present respiratory complications in childhood or adulthood. Molecular analysis of the SLC26A2 gene showed that the patient was compound heterozygote for the Ser157Thr and Arg279Trp substitutions.
The Arg279Trp mutation was inherited from her father. This mutation causes a substitution of arginine for tryptophan at position 279, in a non transmembrane domain of the protein, which would allow a residual activity and an attenuated phenotype of the disease. This mutation is pan-ethnic and it accounts for a thirdof the mutant alleles in the Caucasian population (11).
The Ser157Thr substitution, inherited from her mother, is a novel mutation that has never been reported. We validated its contribution in the development of the disease using PolyPhen 2: the software algorithm predicts that the mutation is probably deleterious because it is found in a conserved region and it is in a transmembrane domain.
There are more than 30 mutations identified in this gene, most of these de novo mutations found in families of different origins. Based on the literature, 50% are missense, 33% are deletions, 10% are nonsense mutations, 5% are splice site mutations and 3 % are insertions (12). In the case of diastrophic dysplasia phenotype, the 5 most common mutations are IVS1 +2 T> C, R279W, V340del, C653S and R178X (13).
There have been made numerous correlations between mutations in the SLC26A2 gene and the severity of the clinical phenotype. DTD patients may be homozygous or compound heterozygous for mutations that allow a residual activity of the transporter protein. However not all cases show this correlation may be because of environmental factors and alternative routes in the recruitment of sulfate that may account for clinical variability (7). Even if functional studies have demonstrated that the Arg279Trp allow a residual function of the protein, it is difficult to assess the contribution of each mutation to the clinical phenotype of our patient since functional studies on the Ser157Thr are not available.
Financiación
Ninguna declarada por los autores.
Conflictos de interés
Ninguno declarado por los autores.
Agradecimientos
Ninguno declarado por los autores.
References
1. Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943-68.
2. Lamy M, Maroteaux P. Diastrophic Nanism. La Presse Médicale. 1960;68: 1977-80.
3. Córdoba-Aguilar H. Naturaleza y Sociedad. Una Introducción a la Geografía. Capítulo 3. Lima: Editorial pontificia Universidad del Perú; 2002.
4. Bonafé L, Mittaz-Crettol L, Ballhausen D, Superti-Furga A. Diastrophic Dysplasia. Gene Reviews. 2004. Available from: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=diastrophic-d.
5. Superti-Furga A. Skeletal dysplasias related to defects in sulfate metabolism. In: Royce P, Steinmann B, editors. Connective Tissue and Its Heritable Disorders, 2 ed. New York: Wiley-Liss, Inc. 2002. 939-60.
6. Hästbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, Hamilton BA, Kusumi K, Trivedi B, Weaver A, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78:1073-87.
7. Macías Gómez N, Megarbané A, Leal Ugarte E, Rodríguez-Rojas L, Barros Núñez P. Diastrophic Dysplasia and Atelostogenesis Type II as Expression of Compound Heterozygosis: First Report of a Mexican Patient and Genotype-Phenotype Correlation. Am J Med Genet A. 2004;129:190-2.
8. Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 2001;17:159-71. Erratum in: HumMutat 2001;18:82.
9. Maeda K, Miyamoto Y, Sawai H, Karniski LP, Nakashima E, Nishimura G, Ikegawa S. A compound heterozygote harboring novel and recurrent DTDST mutations with intermediate phenotype between atelosteogenesis type II and diastrophicdysplasia. Am J Med Genet A. 2006;140:1143-7.
10. Barbosa-Buck CO, Orioli IM, da GraçaDutra M, Lopez-Camelo J, Castilla EE, Cavalcanti DP. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A 2012;158A:1038-45.
11. Conserved domains. Sulfate transporter. [Online]. 2012 [Cited 2012 Dec 23]; Available from: http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?INPUT_TYPE=live&SEQUENCE=NP_000103.2.
12. Dawson PA, Markovich D. Pathogenetics of the Human SLC26 Transporters. Current Medicinal Chemistry. 2005;12:385-96.
13. Diastrophic dysplasia. [Online]. 2012 [Cited 2012 Dec 23]; Available from: http://omim.org/entry/222600.
References
Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943-68.
Lamy M, Maroteaux P. Diastrophic Nanism. La Presse Médicale. 1960;68: 1977-80.
Córdoba-Aguilar H. Naturaleza y Sociedad. Una Introducción a la Geografía. Capítulo 3. Lima: Editorial pontificia Universidad del Perú; 2002.
Bonafé L, Mittaz-Crettol L, Ballhausen D, Superti-Furga A. Diastrophic Dysplasia. Gene Reviews. 2004. Available from: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=diastrophic-d.
Superti-Furga A. Skeletal dysplasias related to defects in sulfate metabolism. In: Royce P, Steinmann B, editors. Connective Tissue and Its Heritable Disorders, 2 ed. New York: Wiley-Liss, Inc. 2002. 939-60.
Hästbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, Hamilton BA, Kusumi K, Trivedi B, Weaver A, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78:1073-87.
Macías Gómez N, Megarbané A, Leal Ugarte E, Rodríguez-Rojas L, Barros Núñez P. Diastrophic Dysplasia and Atelostogenesis Type II as Expression of Compound Heterozygosis: First Report of a Mexican Patient and Genotype-Phenotype Correlation. Am J Med Genet A. 2004;129:190-2.
Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 2001;17:159-71. Erratum in: HumMutat 2001;18:82.
Maeda K, Miyamoto Y, Sawai H, Karniski LP, Nakashima E, Nishimura G, Ikegawa S. A compound heterozygote harboring novel and recurrent DTDST mutations with intermediate phenotype between atelosteogenesis type II and diastrophicdysplasia. Am J Med Genet A. 2006;140:1143-7.
Barbosa-Buck CO, Orioli IM, da GraçaDutra M, Lopez-Camelo J, Castilla EE, Cavalcanti DP. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A 2012;158A:1038-45.
Conserved domains. Sulfate transporter. [Online]. 2012 [Cited 2012 Dec 23]; Available from: http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?INPUT_TYPE=live&SEQUENCE=NP_000103.2.
Dawson PA, Markovich D. Pathogenetics of the Human SLC26 Transporters. Current Medicinal Chemistry. 2005;12:385-96.
Diastrophic dysplasia. [Online]. 2012 [Cited 2012 Dec 23]; Available from: http://omim.org/entry/222600.
How to Cite
APA
ACM
ACS
ABNT
Chicago
Harvard
IEEE
MLA
Turabian
Vancouver
Download Citation
Article abstract page views
Downloads
License
Copyright (c) 2013 Revista de la Facultad de Medicina

This work is licensed under a Creative Commons Attribution 3.0 Unported License.
Copyright
Authors must agree to transfer to the Revista de la Facultad de Medicina the copyright of the articles published in the Journal. The publisher has the right to use, reproduce, transmit, distribute and publish the articles in any form. Authors will not be able to permit or authorize the use of their published paper without the written consent of the Journal.
The letter of copyright transfer and the letter of authorship responsibility must be submitted along with the original paper through the Journal OJS platform. These files are available in https://goo.gl/EfWPdX y https://goo.gl/6zztk4 and must be uploaded in step 4 (supplementary files).
Authors who publish with this journal agree to the following terms:
- Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution License that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.
- Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the journal's published version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgement of its initial publication in this journal.
- Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See The Effect of Open Access).


















