



Publicado
Severe congenital diarrhea secondary to tufting enteropathy. Case report
Diarrea congénita grave secundaria a enteropatía en penacho. Reporte de caso
DOI:
https://doi.org/10.15446/cr.v8n1.90883Palabras clave:
Tufting Enteropathy, Newborn, Infantile Diarrhea, Diarrhea Congenital, Hereditary and Neonatal Diseases and Abnormalities, Intestinal Diseases (en)Diarrea neonatal, Recién nacido, Diarrea infantil, Anomalías congénitas, Enfermedades Intestinales, Enfermedades Raras (es)
Descargas
Introduction: Tufting enteropathy is a rare cause of congenital diarrhea in neonates. It is characterized by the abnormal distribution of epithelial adhesion molecules, which causes enterocytes to shed into the lumen, forming the characteristic tufts.
Case presentation: A 15-day-old female neonate was taken by her parents to the emergency department of a tertiary care hospital due to diarrheal stools she had been experiencing since birth. The patient presented with dehydration, abnormal weight loss, metabolic acidosis, and acute kidney failure. She received treatment with alizapride, loperamide, zinc sulfate, and probiotics, but after 75 days of treatment she was still symptomatic. An upper tract endoscopy and colonoscopy were performed, finding flattening of the villi and lymphoid cells in the lamina propria. However, the symptoms persisted, and she died at the age of ten months. A post-mortem exome sequencing reported tufting enteropathy.
Conclusions. When congenital diarrhea is present, tufting enteropathy should be considered. An early molecular study would allow to evaluate the possibility of performing an intestinal transplant or modifying the treatment to meet the patient’s palliative care needs.
Introducción. La enteropatía en penacho es una causa rara de diarrea congénita en neonatos; esta se caracteriza por una alteración de la adhesión epitelial que ocasiona desprendimiento de enterocitos hacia el lumen y, en consecuencia, forma los característicos penachos. Se describe el caso de una paciente con esta patología.
Presentación del caso. Neonata de 15 días de vida, quien fue llevada por sus padres al servicio de urgencias de un hospital de tercer nivel debido a que desde su nacimiento tuvo deposiciones diarreicas y a causa de esto presentó deshidratación, pérdida de peso, acidosis metabólica e insuficiencia renal aguda. La paciente recibió manejo con alizaprida, loperamida, sulfato de zinc y probióticos, pero a los 75 días de tratamiento continuaba sintomática. Se le practicó una endoscopia de vías digestivas y una colonoscopia que mostraron aplanamiento de las vellosidades e infiltrado de células linfoides en la lámina propia. Los síntomas continuaron y la menor falleció a los 10 meses de nacida. El resultado del exoma post mortem reportó enteropatía en penacho.
Conclusiones. Ante la presencia de diarrea congénita, se debe sospechar de una enteropatía en penacho y considerar el estudio molecular temprano, pues este permite evaluar la posibilidad de realizar un trasplante intestinal o modificar el tratamiento según las necesidades de cuidado paliativo del paciente.
https://doi.org/10.15446/cr.v8n1.90883
Severe congenital diarrhea secondary to tufting enteropathy. Case report
Keywords: Tufting Enteropathy; Newborn; Infantile Diarrhea; Congenital, Hereditary, and Neonatal Diseases and Abnormalities; Intestinal Diseases.
Palabras clave: Diarrea neonatal; Recién nacido; Diarrea infantil; Anomalías congénitas; Enfermedades Intestinales; Enfermedades Raras.
María Angélica Wilches-Cuadros
Universidad del Rosario - School of Medicine and Health Sciences - Department of Pediatrics
- Bogotá, D.C. - Colombia.
Laura González-Hakspiel
Paula Nausa-Suárez
María Paula Fernández
Paula Patiño-Ascencio
Alejandra Manrique-Guerrero
Universidad Autónoma de Bucaramanga - Faculty of Health Sciences - Medical Program - Bucaramanga - Colombia.
Ángela Milena Díaz-Díaz
Universidad Autónoma de Bucaramanga - Faculty of Health Sciences - Medical Program - Bucaramanga - Colombia.
Clínica FOSCAL - Pediatrics Service
- Floridablanca - Colombia
Derly Liseth Castro-Rojas
Centro de Inmunología y genética CIGE -
Medical Genetics Service - Medellín - Colombia
Corresponding author
Ángela Milena Díaz-Díaz. Departamento de Pediatría, Clínica FOSCAL. Floridablanca. Colombia.
Email: adiaz558@unab.edu.co
Received: 12/10/2020 Accepted: 15/03/2021
Resumen
Introducción. La enteropatía en penacho es una causa rara de diarrea congénita en neonatos; esta se caracteriza por una alteración de la adhesión epitelial que ocasiona desprendimiento de enterocitos hacia el lumen y, en consecuencia, forma los característicos penachos. Se describe el caso de una paciente con esta patología.
Presentación del caso. Neonata de 15 días de vida, quien fue llevada por sus padres al servicio de urgencias de un hospital de tercer nivel debido a que desde su nacimiento tuvo deposiciones diarreicas y a causa de esto presentó deshidratación, pérdida de peso, acidosis metabólica e insuficiencia renal aguda. La paciente recibió manejo con alizaprida, loperamida, sulfato de zinc y probióticos, pero a los 75 días de tratamiento continuaba sintomática. Se le practicó una endoscopia de vías digestivas y una colonoscopia que mostraron aplanamiento de las vellosidades e infiltrado de células linfoides en la lámina propia. Los síntomas continuaron y la menor falleció a los 10 meses de nacida. El resultado del exoma post mortem reportó enteropatía en penacho.
Conclusiones. Ante la presencia de diarrea congénita, se debe sospechar de una enteropatía en penacho y considerar el estudio molecular temprano, pues este permite evaluar la posibilidad de realizar un trasplante intestinal o modificar el tratamiento según las necesidades de cuidado paliativo del paciente.
Abstract
Introduction: Tufting enteropathy is a rare cause of congenital diarrhea in neonates. It is characterized by the abnormal distribution of epithelial adhesion molecules, which causes enterocytes to shed into the lumen, forming the characteristic tufts.
Case summary: A 15-day-old female neonate was taken by her parents to the emergency department of a tertiary care hospital due to diarrheal stools she had been experiencing since birth. The patient presented with dehydration, abnormal weight loss, metabolic acidosis, and acute kidney failure. She received treatment with alizapride, loperamide, zinc sulfate, and probiotics, but after 75 days of treatment she was still symptomatic. An upper tract endoscopy and colonoscopy were performed, finding flattening of the villi and lymphoid cells in the lamina propria. However, the symptoms persisted, and she died at the age of ten months. A post-mortem exome sequencing reported tufting enteropathy.
Conclusions. When congenital diarrhea is present, tufting enteropathy should be considered. An early molecular study would allow to evaluate the possibility of performing an intestinal transplant or modifying the treatment to meet the patient’s palliative care needs.
Introduction
Congenital diarrhea is a rare disorder in neonates (1) and differs from acquired diarrhea due to its severity, chronicity, and dependence on nutritional support (2). This condition is caused by epithelial defects and alterations in neuroendocrine differentiation or immune dysregulation, digestion, and nutrient or electrolyte absorption or transport (1). It is classified as severe chronic diarrhea, with a high mortality rate, a difficult diagnosis, and an intractable course (3).
Tufting enteropathy is a rare cause of congenital diarrhea in neonates and is characterized by the abnormal distribution of epithelial adhesion molecules, which causes enterocytes to shed into the lumen, forming the characteristic tufts (3). The incidence of this disease in Europe varies from 1 cases per 50 000 to 100 000 live births; however, a higher incidence is reported in patients of Arab origin (4).
The following is the case of a neonate with tufting enteropathy who presented with intractable diarrhea of infancy and died at 10 months of age despite receiving multiple treatments.
Case presentation
A 15-day-old female neonate from Bucaramanga (Colombia), mestizo, was taken by her parents to the emergency department of a tertiary care hospital because due to explosive, abundant, watery, yellowish liquid stools without mucus or blood since birth. Seven days prior to the consultation, the patient presented postprandial vomiting, irritability, jaundice, and nursing strike, so the parents started supplementation with milk formula at home.
Relevant perinatal history included that this was the mother’s second pregnancy, that she had adequate antenatal care and that the patient was born by vaginal delivery at 40 weeks, weighed 3 245 grams, and Rh incompatibility tests were positive.
The patient was admitted to the emergency room in fair health, with blood pressure of 61/38 mmHg, heart rate of 158 bpm, respiratory rate of 56 rpm, temperature of 35.4°C, oxygen saturation of 90% on room air, glucose level at 232 mg/dL, and weight of 2 800 grams (13% weight loss since birth). Physical examination showed sunken fontanelle, dry oral mucosa, marked intercostal retractions with scarce panniculus adiposus, scaphoid abdomen with hepatomegaly, hypotrophic limbs, and pallor on the elbows; the rest of the examination was normal. Due to the suspicion of late-onset neonatal sepsis, the girl was immediately hospitalized in the neonatal intensive care unit (NICU), where laboratory tests were requested and oxygen support, fluid resuscitation, and empirical antibiotic therapy were initiated.
The test results on admission reported a normal blood count with the following values: hemoglobin: 18.9 g/dL, hematocrit: 56.2%, leukocytes: 18970/mm³, neutrophils: 6499/µL, lymphocytes: 8157/µL and platelets: 81000/mm³. Prerenal acute kidney failure with creatinine of 1.61 mg/dL and BUN of 40.5 mg/dL was also identified. Total bilirubin was 5.91 mg/dL, direct bilirubin was 1.41 mg/dL, and indirect bilirubin was 4.5 mg/dL. C-reactive protein, transaminases, and electrolytes were normal and arterial blood gas levels showed metabolic acidosis with the following vales: pH: 7.29; PaCO2: 22.7 mmHg; PaO2: 103.4 mmHg; HCO3: 10.8 mmol/L; base excess: -13 µmol/L. On the day of admission, two blood cultures were also taken, which were negative after 5 days of incubation.
Once stable in the NICU, enteral feeding was restarted; although it was initially tolerated, with a gradual increase in intake, the patient again presented with abundant liquid stools and hypovolemic shock. She was given alizapride (3 mg/kg/day for 8 days), loperamide (0.08 mg/kg every 12 hours for 8 days), zinc sulfate (5 mL every day for 10 days), and probiotics; however, no improvement was achieved.
After 15 days of hospitalization, the patient was assessed by the genetics department, which requested further tests to rule out inborn errors of carbohydrate metabolism, aminoacidopathies, metabolic acidemias, cystic fibrosis, among others. Seliwanoff’s test was negative, while qualitative amino acid urine tests were positive for sodium nitroprusside. Plasma and urine amino acid quantification tests were also requested, which yielded nonspecific results (Tables 1 and 2).
Table 1 Altered quantitative plasma amino acids
|
Amino acid |
Value obtained (µmol/L) |
Reference value (µmol/L) |
|
Aspartic acid |
22 |
2-14 |
|
Glutamic acid |
230 |
32-185 |
|
Serine |
235 |
83-212 |
|
α-Aminoadipic acid |
15 |
<5 |
|
Glycine |
527 |
103-386 |
|
Sarcosine |
9 |
<5 |
|
Arginine |
179 |
30-147 |
|
γ-Aminobutyric acid |
1 |
<1 |
|
α-Aminobutyric acid |
37 |
4-30 |
|
Phenylalanine |
103 |
31-92 |
|
Ornithine |
206 |
19-139 |
|
Lysine |
427 |
70-258 |
Source: Own elaboration.
Table 2 Altered quantitative amino acids in urine.
|
Amino acid |
Value obtained (µmol/µol creatinine) |
Reference values (µmol/µol creatinine) |
|
Alanine |
431 |
27-313 |
|
Glycine |
2988.31 |
133-894 |
|
Valine |
56.56 |
3-43 |
|
Isoleucine |
23.57 |
2-21 |
|
Threonine |
287.52 |
12-145 |
|
Serine |
638.67 |
34-329 |
|
Phenylalanine |
44.78 |
7-42 |
|
α-Aminoadipic acid |
160.26 |
7-110 |
|
Ornithine |
120.19 |
2-19 |
|
Lysine |
549.11 |
4-80 |
|
Histidine |
841.35 |
69-392 |
Source: Own elaboration.
At one week of age, the patient underwent an abdominal ultrasound, followed by a brain magnetic resonance imaging and echocardiogram two weeks later, all of which were normal. There were no cataracts in the fundus and the pilocarpine iontophoresis test (at three different times) was normal.
The patient was also assessed by the pediatric nephrology service at one month of life, which diagnosed high anion gap metabolic acidosis and negative urinary anion gap and suggested a high probability of an inborn error of metabolism rather than tubular acidosis.
After a 75-day hospital stay, during which she was readmitted to the NICU three times due to hypovolemic shock caused by diarrhea following the administration of various nutritional formulas, the patient, who was already 3 months old at that time, weighed 3.59kg and measured 52cm (standard deviations for weight-for-age, height-for-age and weight-for-height were -3.99, -4.03, and -0.61, respectively), which indicated failure to thrive. Additionally, chronic diarrhea and infectious episodes persisted.
At that point, she was evaluated by the pediatric gastroenterology service, which diagnosed intractable diarrhea of infancy and started treatment with ondansetron (0.15 mg/kg/dose for 5 days) and requested a pancreatic enzyme replacement therapy for 2 months (lipase), but no improvement was observed. Endoscopy and colonoscopy were also performed, identifying flattening of the villi in the bulb, in the second portion of the duodenum, and in the last 5 centimeters of the ileum mucosa. Biopsies were taken during these last two examinations, which were processed with electron microscopy, revealing ileum with lymphoid cells in the lamina propria, without microscopic abnormalities. Based on the results, ultrastructural changes suggestive of microvillous inclusion disease or altered lipid transport were ruled out.
As previously stated, the patient developed hypovolemic shock several times as a result of diarrhea, which required fluid resuscitation, prolonged total parenteral nutrition, and gastrostomy feeding. A new amino acid test showed altered aminoaciduria, as well as aminoacidemia with increased oligosaccharides in urine, for which exome sequencing was indicated.
At 10 months of age, vascular access was lost and the only treatment option available was a transhepatic catheter, which her parents rejected due to due to the high morbidity associated with the procedure. The day after this event, the child died without a known diagnosis, but 2 months later, the exome sequencing report indicated a mutation and deletion of the EPCAM gene (Table 3), which encodes an epithelial adhesion molecule expressed in the cell membrane. It has been reported that these pathogenic variants cause tufting enteropathy with subsequent diarrhea. Figure 1 shows the clinical course and management of the patient during her hospital stay.
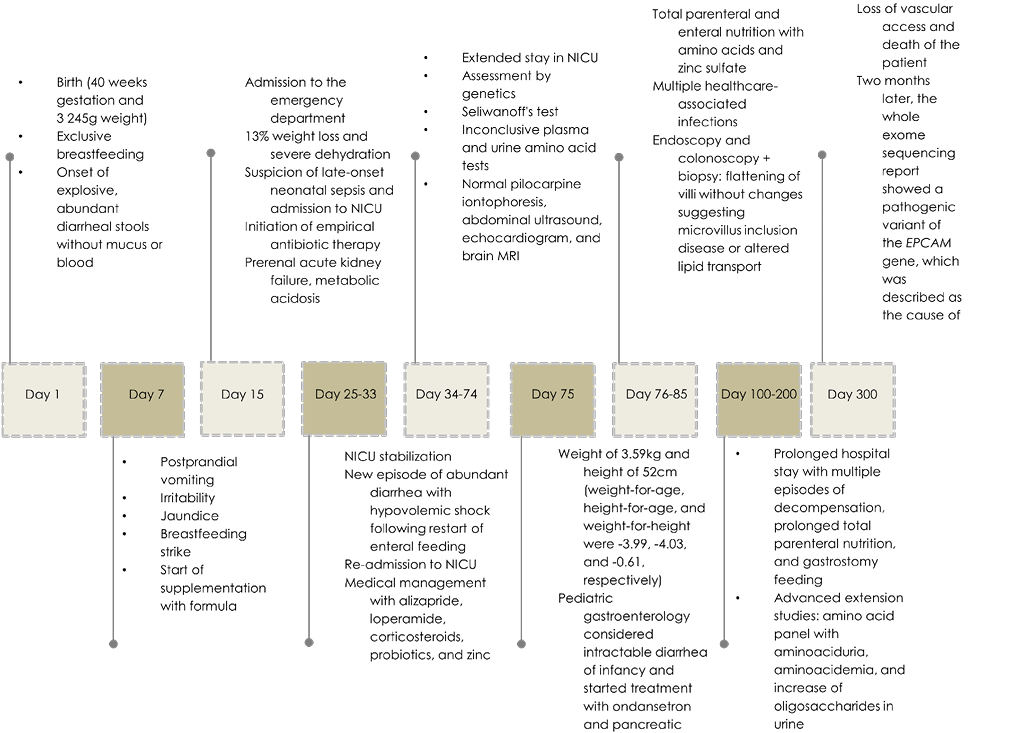
Figure 1 Timeline. NICU: neonatal intensive care unit; NMR: nuclear magnetic resonance; SD: standard deviation.
Source: Own elaboration.
Table 3 Exome sequencing.
|
Gene |
Variant |
Zygosity |
Inheritance |
MAF (%) |
Prediction In silico |
Classification |
||
|
Case |
Mother |
Father |
||||||
|
EPCAM |
c.492-3C>G;p.? |
het. |
- |
het. |
AR, AD |
- |
Affecting splicing |
Unknown meaning |
|
EPCAM |
Del.exon 1-3 |
het. |
het. |
- |
AR, AD |
- |
- |
Probably deleterious |
HET: heterogeneous; AR: autosomal recessive inheritance; AD: autosomal dominant disorder; MAF: minor allele frequency.
Source: Own elaboration.
Discussion
Tufting enteropathy, also known as intestinal epithelial dysplasia, is an autosomal recessive genetic disorder that is usually caused by mutations in the EPCAM gene, a type I transmembrane surface glycoprotein antigen expressed on the basolateral membrane of multiple epithelial and plasma cells (5). This gene is located in the 2p21 locus, and when it mutates, it causes abnormal development of the intestinal mucosa (6), which is caused by changes in epithelial adhesion molecules and causes enterocytes to shed into the lumen, resulting in the formation of tufts (7). This disease occurs during the first few months of life and manifests with chronic watery diarrhea that persists despite intestinal rest (8) and feeding with breast milk or formula (5). It usually causes a deterioration in the patient’s growth and is related to other alterations involving the epithelium (9).
Although mutations in the SPINT2 gene have also been described, the most involved gene, as in the case presented here, is EPCAM (10). It encodes a typical adhesion molecule that is connected to the actin cytoskeleton, is directly associated with the tight junction protein claudin-7 and facilitates intestinal barrier formation by recruiting claudins in cell-to-cell junctions (5). Most mutations are located in exons 3, 4, or 5 and lack the extracellular domain or the transmembrane domain. Some deletions in the gene or exon have also been reported, which are related to the degree of involvement caused by the disease (10).
Mutations associated with tufting enteropathy appear to lead to the loss of cell-surface EPCAM protein (4), which is consistent with the immunopathological findings of the present case. Since this is an autosomal recessive disorder, mutations in both alleles are required to express the disease. The exome report in the present case showed a variant of uncertain significance and another probably pathogenic, which, in addition to the clinical findings, allow confirming diagnosis of this condition.
The characteristic feature of tufting enteropathy is the presence of focal epithelial “tufts” composed of tightly packed enterocytes that surround the apical plasma membrane and result in a tear configuration of the affected epithelial cell (7). Hypoplasia and total or partial villi atrophy are commonly associated complications. Moreover, abnormalities of the basement membrane, accumulation of secretory granules in the apical cytoplasm of enterocytes without inflammatory infiltrate, crypt hyperplasia, and normal or slightly increased density of inflammatory cells in the lamina propria may be observed (5,11,12).
In patients with this disease, electron microscopy identifies pathognomonic changes such as the presence of intracytoplasmic vacuoles with microvilli and absent or abnormal microvilli at the luminal border (13). In cases of tufting enteropathy, the number of intraepithelial lymphocytes is not increased, unlike what happens in celiac disease or autoimmune enteropathy (8). In addition, sometimes the characteristic tufts may be absent in early childhood, as in the case of the patient reported here, so a biopsy is required to establish the correct diagnosis (6,9).
The differential diagnoses for diseases with persistent diarrhea and villous atrophy that should be ruled out include congenital chloride diarrhea, congenital sodium diarrhea, and microvillous inclusion disease. In the latter, besides atrophy, positive periodic acid-Schiff (PAS) stained granules are observed in the apical surface of enterocytes with atrophic bands that represent microvillus inclusion in histology (9,12,14). The first two differ in that there is usually no history of polyhydramnios in tufting enteropathy, but there is a history of consanguinity and no electrolyte alterations in blood and feces (9). Similarly, tufting enteropathy differs from glucose-galactose malabsorption in that the latter improves with one hour of fasting and symptoms can be managed through diet modification (9,14), and from autoimmune enteropathy in that it does not respond to immunosuppressive therapy (9).
Tufting enteropathy rapidly threatens the patient’s life due to dehydration and the water-electrolyte disorders it causes (9), as in the reported case, which resulted in the patient’s death due to metabolic decompensation. In patients with this disease, administering elemental formulas or hydrolyzed proteins exacerbates diarrhea, so they quickly become dependent on permanent parenteral feeding.
Regarding the management of tufting enteropathy, intestinal transplantation is indicated in patients with liver disease associated with intestinal failure with recurrent catheter-related sepsis (indication still under debate) and thrombosis of two or more central vascular access sites (15). Genetic counseling should be offered since this condition is related to the presence of parental consanguinity or heterozygous carrier status in asymptomatic parents (9). In cases in which transplant is contraindicated, long-term palliative care with home parenteral nutrition is prescribed (9).
Finally, molecular studies are recommended because they have improved diagnostic techniques and reduced the performance of invasive and costly procedures (5).
Among the strengths of this case report, it is noted that a pathogenic approach was taken using different advanced extension tests, which clarified the various etiological possibilities and, eventually, established a diagnosis of the disease. The patient also received a multidisciplinary treatment, allowing for a comprehensive approach to her disease. On the contrary, limitations include the scarcity of data on the subject due to the few cases reported and the delay in obtaining the results of advanced studies, which led to a late diagnosis that did not allow making interventions aimed at modifying the prognosis and/or improving the patient’s survival.
Conclusion
Congenital diarrhea is a rare disease that usually appears in the neonatal period. It is severe, chronic and difficult to treat, and it predisposes to multiple complications due to the nutritional involvement of the patient, resulting in high morbidity and mortality. Furthermore, due to the rarity and scarcity of cases reported in the literature, knowledge about this disorder is limited, as is its diagnosis.
In cases of refractory congenital diarrhea, genetic defects in enterocytes should be suspected early in order to perform a molecular analysis, as these new diagnostic methods are more efficient and have reduced invasive and costly procedures.
In this sense, in the presence of congenital diarrhea, tufting enteropathy should be suspected, and an early molecular study should be considered because it allows evaluating the possibility of performing an intestinal transplant or modifying the treatment based on the patient’s palliative care needs.
To date, no specific treatment for tufting enteropathy has been described; however, a timely diagnosis allows for the possibility of performing, if indicated, an intestinal transplant on an experimental basis or providing timely therapy to avoid complications such as advanced life support measures and prolonged suffering of patients and their families. Genetic counseling should also be offered to parents because this is an autosomal recessive disease.
Ethical considerations
Informed consent was obtained from the mother of the patient, who authorized the publication of the medical history data for the preparation of this case report.
Conflicts of interest
None stated by the authors.
Funding
None stated by the authors.
Acknowledgments
To the Pediatrics Seedbed of the Universidad Autónoma de Bucaramanga, which brought us together for this purpose.
References
1.Guarino A, Lo Vecchio A, Berni Canani R. Chronic diarrhoea in children. Best Pract Res Clin Gastroenterol. 2012;26(5):649-61. https://doi.org/f2hdz6.
2.O’Connell AE, Zhou F, Shah MS, Murphy Q, Rickner H, Kelsen J, et al. Neonatal-Onset Chronic Diarrhea Caused by Homozygous Nonsense WNT2B Mutations. Am J Hum Genet. 2018;103(1):131-7. https://doi.org/gdxdwc.
3.Russo P. Enteropathies of infancy and childhood. Adv Pediatr. 2013;60(1):217-61. https://doi.org/hgxg.
4.Tan QK-G, Cardona DM, Rehder CW, McDonald MT. Identification of EPCAM mutation: clinical use of microarray. Clin Case Reports. 2017;5(6):980-5. https://doi.org/hgxh.
5.Cai C, Chen Y, Chen, X, Ji F. Tufting enteropathy: a review of clinical and histological presentation, etiology, management, and outcome. Gastroenterol Res Pract. 2020;2020:5608069. https://doi.org/hgxj.
6.Ensari A, Kelsen J, Russo P. Newcomers in paediatric GI pathology: childhood enteropathies including very early onset monogenic IBD. Virchows Arch. 2018;472(1):111-23. https://doi.org/gc72tb.
7.Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: Defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr. 2004;38(1):16-26. https://doi.org/c88xff.
8.Kahvecioğlu D, Yıldız D, Kılıç A, İnce-Alkan B, Erdeve Ö, Kuloğlu Z, et al. A rare cause of congenital diarrhea in a Turkish newborn: Tufting enteropathy. Turk J Pediatr. 2014;56(4):440-3.
9.Goulet O, Salomon J, Ruemmele F, Patey-Mariaud de Serres N, Brousse N. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;2:20. https://doi.org/fjktzq.
10.Das B, Sivagnanam M. Congenital Tufting Enteropathy: Biology, Pathogenesis and Mechanisms. J Clin Med. 2021;10(1):19. https://doi.org/hgxk.
11.Schoen K, Puchi A, González I, Torres MT, Espinosa R, González R. Enfermedad por inclusión microvellositaria como causa de diarrea congénita severa. Caso clínico. Rev Chil Pediatr. 2017;88(5):662-7. https://doi.org/hgxm.
12.Bosaleh A, Contreras M, García de Dávila MT. Enteropatía en penacho: reporte de un caso, metodología de estudio de la biopsia y diagnósticos diferenciales. Acta Gastroenterol Latinoam. 2015;45(1):65-9.
13.Khubchandani SR, Vohra P, Chitale AR, Sidana P. Microvillous inclusion disease - an ultrastructural diagnosis: with a review of the literature. Ultrastruct Pathol. 2011;35(2):87-91. https://doi.org/b42gdd.
14.Thiagarajah JR, Kamin DS, Acra S, Goldsmith JD, Roland JT, Lencer WI, et al. Advances in Evaluation of Chronic Diarrhea in Infants. Gastroenterology. 2018;154(8):2045-2059.e6. https://doi.org/gdcx66.
15.Sudan D. The Current State of Intestine Transplantation: Indications, Techniques, Outcomes and Challenges. Am J Transplant. 2014;14(9):1976-84. https://doi.org/f6kh56.
Referencias
Guarino A, Lo Vecchio A, Berni Canani R. Chronic diarrhoea in children. Best Pract Res Clin Gastroenterol. 2012;26(5):649-61. https://doi.org/f2hdz6.
O’Connell AE, Zhou F, Shah MS, Murphy Q, Rickner H, Kelsen J, et al. Neonatal-Onset Chronic Diarrhea Caused by Homozygous Nonsense WNT2B Mutations. Am J Hum Genet. 2018;103(1):131-7. https://doi.org/gdxdwc.
Russo P. Enteropathies of infancy and childhood. Adv Pediatr. 2013;60(1):217-61. https://doi.org/hgxg.
Tan QK-G, Cardona DM, Rehder CW, McDonald MT. Identification of EPCAM mutation: clinical use of microarray. Clin Case Reports. 2017;5(6):980-5. https://doi.org/hgxh.
Cai C, Chen Y, Chen, X, Ji F. Tufting enteropathy: a review of clinical and histological presentation, etiology, management, and outcome. Gastroenterol Res Pract. 2020;2020:5608069. https://doi.org/hgxj.
Ensari A, Kelsen J, Russo P. Newcomers in paediatric GI pathology: childhood enteropathies including very early onset monogenic IBD. Virchows Arch. 2018;472(1):111-23. https://doi.org/gc72tb.
Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: Defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr. 2004;38(1):16-26. https://doi.org/c88xff.
Kahvecioğlu D, Yıldız D, Kılıç A, İnce-Alkan B, Erdeve Ö, Kuloğlu Z, et al. A rare cause of congenital diarrhea in a Turkish newborn: Tufting enteropathy. Turk J Pediatr. 2014;56(4):440-3.
Goulet O, Salomon J, Ruemmele F, Patey-Mariaud de Serres N, Brousse N. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;2:20. https://doi.org/fjktzq.
Das B, Sivagnanam M. Congenital Tufting Enteropathy: Biology, Pathogenesis and Mechanisms. J Clin Med. 2021;10(1):19. https://doi.org/hgxk.
Schoen K, Puchi A, González I, Torres MT, Espinosa R, González R. Enfermedad por inclusión microvellositaria como causa de diarrea congénita severa. Caso clínico. Rev Chil Pediatr. 2017;88(5):662-7. https://doi.org/hgxm.
Bosaleh A, Contreras M, García de Dávila MT. Enteropatía en penacho: reporte de un caso, metodología de estudio de la biopsia y diagnósticos diferenciales. Acta Gastroenterol Latinoam. 2015;45(1):65-9.
Khubchandani SR, Vohra P, Chitale AR, Sidana P. Microvillous inclusion disease - an ultrastructural diagnosis: with a review of the literature. Ultrastruct Pathol. 2011;35(2):87-91. https://doi.org/b42gdd.
Thiagarajah JR, Kamin DS, Acra S, Goldsmith JD, Roland JT, Lencer WI, et al. Advances in Evaluation of Chronic Diarrhea in Infants. Gastroenterology. 2018;154(8):2045-2059.e6. https://doi.org/gdcx66.
Sudan D. The Current State of Intestine Transplantation: Indications, Techniques, Outcomes and Challenges. Am J Transplant. 2014;14(9):1976-84. https://doi.org/f6kh56.
Cómo citar
APA
ACM
ACS
ABNT
Chicago
Harvard
IEEE
MLA
Turabian
Vancouver
Descargar cita
Licencia

Esta obra está bajo una licencia internacional Creative Commons Atribución 4.0.
Los autores al someter sus manuscritos conservarán sus derechos de autor. La revista tiene el derecho del uso, reproducción, transmisión, distribución y publicación en cualquier forma o medio. Los autores no podrán permitir o autorizar el uso de la contribución sin el consentimiento escrito de la revista.
El Formulario de Divulgación Uniforme para posibles Conflictos de Interés y los oficios de cesión de derechos y de responsabilidad deben ser entregados junto con el original.
Aquellos autores/as que tengan publicaciones con esta revista, aceptan los términos siguientes:
Los autores/as conservarán sus derechos de autor y garantizarán a la revista el derecho de primera publicación de su obra, el cual estará simultáneamente sujeto a la Licencia de reconocimiento de Creative Commons 4.0 que permite a terceros compartir la obra siempre que se indique su autor y su primera publicación en esta revista.
Los autores/as podrán adoptar otros acuerdos de licencia no exclusiva de distribución de la versión de la obra publicada (p. ej.: depositarla en un archivo telemático institucional o publicarla en un volumen monográfico) siempre que se indique la publicación inicial en esta revista.
Se permite y recomienda a los autores/as difundir su obra a través de Internet (p. ej.: en archivos telemáticos institucionales o en su página web) antes y durante el proceso de envío, lo cual puede producir intercambios interesantes y aumentar las citas de la obra publicada. (Véase El efecto del acceso abierto).























