



Publicado
Síndrome de Waardenburg tipo 1 en gemelos monocigóticos y su familia
Type 1 Waardenburg Syndrome in Monozygotic Twins and their Family
DOI:
https://doi.org/10.15446/revfacmed.v64n2.50290Palabras clave:
Síndrome de Waardenburg, Cresta Neural, Anomalías congénitas (es)Waardenburg Syndrome, Neural Crest, Congenital Abnormalities (en)
El síndrome de Waardenburg (SW) es un trastorno genético poco frecuente con una incidencia de 1 por 40000 habitantes. Es originado por mutaciones en múltiples genes como PAX3, MITF, SNAI2 y SOX10; estas alteraciones genéticas ocasionan anomalías en el desarrollo de los tejidos derivados de las células de la cresta neural y producen hallazgos fenotípicos característicos como iris de color azul claro o heterocromía del iris, poliosis, sordera neurosensorial, entre otros.
El objetivo de este artículo es reportar a la literatura un caso poco frecuente de gemelos monocigóticos con hallazgos clínicos típicos de síndrome de Waardenburg tipo 1 con fenotipo diferente entre ellos, su madre y su abuela. Aquí también se establece la importancia del índice W en el diagnóstico y clasificación de este síndrome.
Los hallazgos aquí reportados muestran la variabilidad de las manifestaciones fenotípicas del síndrome de Waardenburg tipo 1 dentro de una familia y especialmente en gemelos monocigóticos, lo que se ha explicado por la expresión variable de genes específicos o por la interacción de ellos con genes modificadores. Cabe resaltar que los gemelos fueron expuestos a alcohol en el primer trimestre del embarazo, por lo cual se propone que la expresión variable del SW fue influenciada por exposiciones a agentes medioambientales.
Waardenburg Syndrome is a rare genetic disorder with an incidence of 1 in 40.000 individuals. It is caused by mutations in multiple genes such as PAX3, MITF, SNAI2 and SOX10. These genetic alterations cause abnormal development of tissues derived from neural crest cells and produce phenotypic characteristic findings as light blue iris or iris heterochromia, poliosis and sensorineural hearing loss, among others.
The aim of this article is to report to the literature a rare case of monozygotic twins with typical clinical findings of type 1 Waardenburg Syndrome with different phenotype between them, including their mother and grandmother. The use of W index to identify cantorum dystopia and to classify the cases according to the four types of Waardenburg syndrome is explained.
The findings reported here show the variability of phenotypic manifestations of type 1 Waardenburg Syndrome within a family and particularly in monozygotic twins, which is explained by the variable expression of specific genes or the interaction of these with modifier genes. Given the fetal exposure of the twins to alcohol, it is proposed that the variable expression of Waardenburg Syndrome would be influenced by exposure to environmental agents.
REPORTE DE CASO
DOI: https://doi.org/10.15446/revfacmed.v64n2.50290
Síndrome de Waardenburg tipo 1 en gemelos monocigóticos y su familia
Type 1 Waardenburg Syndrome in Monozygotic Twins and their Family
Gustavo Andrés Duque1 • Julián Ramírez-Cheyne2 • Wilmar Saldarriaga-Gil2
Recibido: 19/05/2015 Aceptado: 09/09/2015
1 Universidad del Valle - Facultad de Salud - Medicina y Cirugía - Cali - Colombia.
2 Universidad del Valle - Facultad de Salud - Departamento Morfología - Grupo de investigación MACOS - Cali - Colombia.
Correspondencia: Wilmar Saldarriaga-Gil. Departamento de Morfología, Universidad del Valle. Calle 4B No. 36-00, edificio 116, oficina 30. Teléfono: +57 2 5185627, extensión: 4030. Cali. Colombia. Correo electrónico: wilmar.saldarriaga@correounivalle.edu.co.
| Resumen |
El síndrome de Waardenburg (SW) es un trastorno genético poco frecuente con una incidencia de 1 por 40000 habitantes. Es originado por mutaciones en múltiples genes como PAX3, MITF, SNAI2 y SOX10; estas alteraciones genéticas ocasionan anomalías en el desarrollo de los tejidos derivados de las células de la cresta neural y producen hallazgos fenotípicos característicos como iris de color azul claro o heterocromía del iris, poliosis, sordera neurosensorial, entre otros.
El objetivo de este artículo es reportar a la literatura un caso poco frecuente de gemelos monocigóticos con hallazgos clínicos típicos de síndrome de Waardenburg tipo 1 con fenotipo diferente entre ellos, su madre y su abuela. Aquí también se establece la importancia del índice W en el diagnóstico y clasificación de este síndrome.
Los hallazgos aquí reportados muestran la variabilidad de las manifestaciones fenotípicas del síndrome de Waardenburg tipo 1 dentro de una familia y especialmente en gemelos monocigóticos, lo que se ha explicado por la expresión variable de genes específicos o por la interacción de ellos con genes modificadores. Cabe resaltar que los gemelos fueron expuestos a alcohol en el primer trimestre del embarazo, por lo cual se propone que la expresión variable del SW fue influenciada por exposiciones a agentes medioambientales.
Palabras clave: Síndrome de Waardenburg; Cresta Neural; Anomalías congénitas (DeCS).
Duque GA, Ramírez-Cheyne J, Saldarriaga-Gil W. Síndrome de Waardenburg tipo 1 en gemelos monocigóticos y su familia. Rev. Fac. Med. 2016;64(2):365-71. Spanish. doi: https://doi.org/10.15446/revfacmed.v64n2.50290.
Abstract
Waardenburg Syndrome is a rare genetic disorder with an incidence of 1 in 40.000 individuals. It is caused by mutations in multiple genes such as PAX3, MITF, SNAI2 and SOX10. These genetic alterations cause abnormal development of tissues derived from neural crest cells and produce phenotypic characteristic findings as light blue iris or iris heterochromia, poliosis and sensorineural hearing loss, among others.
The aim of this article is to report to the literature a rare case of monozygotic twins with typical clinical findings of type 1 Waardenburg Syndrome with different phenotype between them, including their mother and grandmother. The use of W index to identify cantorum dystopia and to classify the cases according to the four types of Waardenburg syndrome is explained.
The findings reported here show the variability of phenotypic manifestations of type 1 Waardenburg Syndrome within a family and particularly in monozygotic twins, which is explained by the variable expression of specific genes or the interaction of these with modifier genes. Given the fetal exposure of the twins to alcohol, it is proposed that the variable expression of Waardenburg Syndrome would be influenced by exposure to environmental agents.
Keywords: Waardenburg Syndrome; Neural Crest; Congenital Abnormalities (MeSH).
Duque GA, Ramírez-Cheyne J, Saldarriaga-Gil W. [Type 1 Waardenburg Syndrome in Monozygotic Twins and their Family]. Rev. Fac. Med. 2016;64(2): 365-71. Spanish. doi: https://doi.org/10.15446/revfacmed.v64n2.50290.
Introducción
El síndrome de Waardenburg (SW) es un trastorno genético raro que tiene una incidencia de 1 por 40000 individuos (1) y fue descrito por primera vez en 1951 por Petrus Johannes Waardenburg (2). Es originado por mutaciones en múltiples genes como PAX3, MITF, SNAI2, SOX10, entre otros (3); estas mutaciones ocasionan anomalías en el desarrollo de los tejidos derivados de las células de la cresta neural (4,5).
Las manifestaciones clínicas principales del SW son la distopia cantorum, hipopigmentación del iris incluyendo la heterocromía ocular, mechón de pelo blanco denominado poliosis y pérdida de la audición neurosensorial, entre otros (1,4). La expresión fenotípica es muy variable, razón por la cual el Consorcio Internacional para el Estudio del Síndrome de Waardenburg desarrolló criterios clínicos diagnósticos (6). Este síndrome es clasificado en cuatro tipos que representan las manifestaciones clásicas pero se diferencian por variaciones genotípicas y fenotípicas (7). En el tipo 1 se afecta el gen PAX3 y el en el tipo 2 los genes MITF, SNAI2 y SOX10, ambos tienen presencia de distopia cantorum; en el tipo 3 se encuentran alteraciones músculoesqueléticas y el gen alterado es el PAX3; finalmente, el tipo 4 presenta síndrome de Hirschsprung y los genes afectados son SOX10, EDN3 y EDNRB.
El objetivo de este artículo es reportar a la literatura un caso muy poco frecuente de gemelos monocigóticos con hallazgos clínicos típicos de SW tipo 1 con fenotipo diferente; además, aportar a la comunidad médica herramientas para sospechar, diagnosticar y clasificar clínicamente el SW.
La madre de los pacientes aceptó y firmó el consentimiento informado para toma de fotografías y uso de datos de las historias clínicas para esta publicación.
Descripción de los casos
Gemelos de sexo masculino fueron llevados por su madre y abuela materna al Servicio de Dismorfología y Genética Médica del Hospital Universitario del Valle “Evaristo García” de Santiago de Cali, Colombia (Figura 1) cuando tenían 20 meses de edad. Hijos de padres no consanguíneos. La madre se embarazó de los gemelos a los 17 años, con único antecedente obstétrico de un aborto espontaneo; en este embarazo el único hallazgo relevante fue el consumo de alcohol durante el primer trimestre. En las ecografías obstétricas se reportó embarazo gemelar monocoriónico biamniótico y se observó labio y paladar fisurado en ambos gemelos sin más anomalías. Se realizó cesárea a las 35 semanas de gestación y se requirió hospitalización neonatal por exposición a ruptura prematura de membranas. En la nota quirúrgica del obstetra se ratificó que el embarazo era monocorial biamniótico, por lo cual se concluyó que eran gemelos monocigóticos.
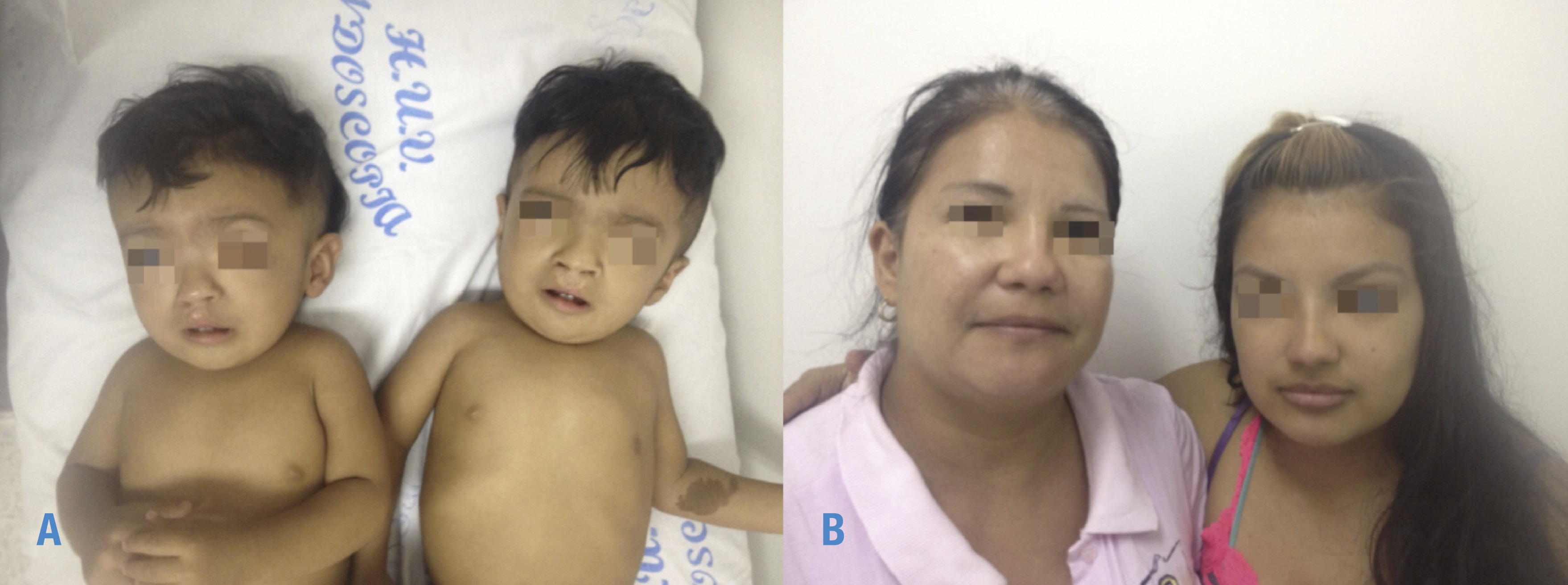
Figura 1. Fotos de gemelos, madre y abuela con síndrome de Waardeburg. A) Izquierda: gemelo 1 con cicatriz de corrección quirúrgica de labio fisurado derecho, derecha: gemelo 2 con cicatriz de corrección quirúrgica de labio fisurado bilateral y mácula hiperpigmentada en antebrazo izquierdo; B) izquierda: abuela materna con poliosis, derecha: madre con tinte en cabello cubriendo la poliosis. Fuente: Documento obtenido durante la realización del estudio.
A continuación se hace la descripción de los hallazgos clínicos relevantes en gemelos, madre y abuela al momento de la consulta; el índice W, IW (8), se calculó reemplazando los valores en milímetros de la distancia intercantal interna (A), la distancia inter pupilar (B) y la distancia intercantal externa (C) con las siguientes formulas:
Calcular X=(2A-(0.2119C+3.909))/C
Calcular Y=(2A-(0.2479B+3.909))/B
Calcular W=X+Y+A/B
Gemelo 1: Talla 76cm, desviación estándar (σ) -3.42; peso 8kg, σ -3.40; perímetro cefálico (PC) 46cm, σ -1.47; facies especiales: heterocromía del iris, ojo derecho azul e izquierdo café; estrabismo convergente en ojo derecho; cejas con el tercio medial amplio y sinofris insinuado (Figura 2); puente nasal ancho, distancia intercantal interna (A): 35mm >p97, distancia interpupilar (B): 55mm >p97 y distancia intercantal externa (C): 90mm >p97, con un índice W de 2.11; cicatriz de corrección quirúrgica de labio fisurado derecho. No se encontraron alteraciones en extremidades, los hitos del desarrollo psicomotor para la edad fueron cumplidos adecuadamente, potenciales auditivos no mostraron hipoacusia y no presenta estreñimiento.
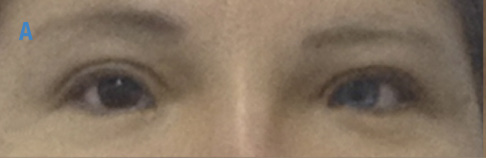

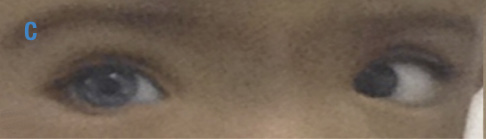
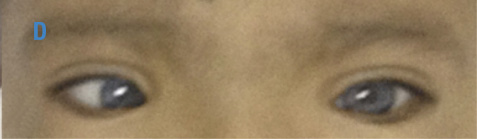
Figura 2. Foto de globo ocular y tejidos blandos en región periorbitaria de los gemelos, madre y abuela con síndrome de Waardenburg. A) Abuela materna con heterocromía del iris, estrabismo convergente ojo izquierdo y distopía cantorum; B) Madre con ojos azules brillantes y distopía cantorum; C) Gemelo 1 con heterocromía del iris, estrabismo convergente ojo izquierdo y distopía cantorum; D) Gemelo 2 con ojos azules brillantes, estrabismo convergente derecho y distopía cantorum. Fuente: Documento obtenido durante la realización del estudio.
Gemelo 2: Talla 75cm, σ -3.77; peso 7kg, σ -4.058; PC 45cm, σ -2.21; facies especiales: ojos color azul brillantes, estrabismo convergente izquierdo (Figura 2); puente nasal ancho y prominente, distancia intercantal interna (A): 40mm > p97, distancia interpupilar (B): 55mm >p97 y distancia intercantal externa (C): 90mm >p97, con un índice W de 2.49; cicatriz de corrección quirúrgica de labio fisurado bilateral y paladar hendido; macula hiperpigmentada en antebrazo izquierdo con limites irregulares de 7cm por 4cm aproximadamente. No se encontraron alteraciones en extremidades, los hitos del desarrollo psicomotor para la edad fueron cumplidos adecuadamente, potenciales auditivos no mostraron hipoacusia y no presenta estreñimiento.
Madre: mechón de cabello blanco en región central que se cubre con tinte; ojos de color azul brillante, puente nasal ancho (Figura 2); distancia intercantal interna (A): 45mm >p97, distancia interpupilar (B): 65mm >p97 y distancia intercantal externa (C): 110mm >p97, con un índice W 2.333; hipoacusia severa izquierda de origen neurosensorial y no presenta estreñimiento.
Abuela: mechón de cabello blanco en región central, heterocromía del iris, ojo derecho café, ojo izquierdo azul y estrabismo convergente ojo izquierdo (Figura 2); puente nasal ancho y prominente; distancia intercantal interna (A): 40mm >p97, distancia interpupilar (B): 65 mm >p97 y distancia intercantal externa (C): 110mm >p97, con un índice W de 2.297; no presenta hipoacusia ni estreñimiento.
Como antecedentes familiares cabe resaltar que el padre de la abuela materna presentó heterocromía del iris, poliosis e hipoacusia derecha; de igual forma, un familiar del padre presentó labio fisurado unilateral y paladar hendido (Figura 3). Dados los hallazgos clínicos de los gemelos y su historia familiar se diagnosticó SW tipo 1. En la Figura 3 se evidencia el patrón de herencia autosómico dominante del SW presente en las generaciones I, II, III y IV de la familia materna y la presencia de un caso de labio y paladar hendido aislado en la familia paterna.

Figura 3. Heredograma. Fuente: Elaboración propia.
Discusión
El SW es una enfermedad genética clasificada dentro de las neurocristopatías (9) y originada por alteraciones en genes cuyos derivados proteicos intervienen de forma variable en la diferenciación de los tejidos derivados de las células de la cresta neural como los melanocitos, células mesodérmicas que generan la eminencia frontonasal, células neurosensoriales del oído interno, entre otras. Lo anterior explica la pleiotropía y la expresividad variable del SW, lo cual se evidencia en que las características fenotípicas de los casos presentan diferentes grados de afectación tanto en los trastornos pigmentarios como en las alteraciones faciales y la hipoacusia neurosensorial (Tabla 1) (4,10).
Tabla 1. Genes mutados, herencia y características clínicas diferenciales en los cuatro tipos del síndrome de Waardenburg.
|
Tipo de SW |
Hallazgo clínico distintivo |
Genes mutados |
Locus |
Herencia |
|
1 |
Distopia cantorum presente |
PAX3 |
2q36.1 |
AD |
|
2 |
Distopia cantorum ausente |
MITF, SNAI2, SOX10. |
3p13, 8q1.21, 22q13.1. |
AD |
|
3 |
Alteraciones músculo esqueléticas |
PAX3 |
2q36.1 |
AD y AR |
|
4 |
Enfermedad de Hirschprung |
SOX10, EDN3, EDNRB |
22q13.1, 20q13.32, 13q22.3. |
Principalmente AR |
Fuente: Elaboración propia.
Mutaciones en varios genes con locus en diferentes cromosomas se han establecido como causantes de los cuatro tipos de SW, que además se diferencian en sus características fenotípicas y patrones de herencia (Tabla 1) (11,12).
Las mutaciones en el gen PAX3, que presentan herencia autosómica dominante, son responsables de los tipos 1 y 3 del SW (12,25). Este gen juega un importante rol en la embriogénesis, codifica un factor de transcripción que se une al ADN y es fundamental en el mantenimiento de las células madre pluripotenciales, la especificación de linajes celulares, proliferación, migración, apoptosis e inhibición de la diferenciación terminal, especialmente expresado en los derivados de la cresta neural, incluyendo los melanocitos en donde su expresión es determinante para una adecuada migración y diferenciación (13,14,15).
El impacto que tienen las mutaciones en el gen PAX3 sobre el fenotipo aún no se encuentra totalmente claro, dado que la expresión fenotípica es extremadamente variable, incluso dentro de una misma familia como la descrita en este reporte. Aquí encontramos tres generaciones examinadas con un fenotipo diferente, dado que la madre de los gemelos tenia hipoacusia, pero no sus hijos ni su madre; para lo cual se ha propuesto que existe una posible interacción con genes modificadores que explicaría que a pesar de ser la misma mutación no tengan iguales hallazgos clínicos (13).
Sin embargo, en esta familia se encontró un gemelo monocigótico con heterocromía del iris y labio fisurado unilateral y el otro con los ojos azules brillantes, labio fisurado bilateral y paladar hendido, lo cual contradice la hipótesis de genes modificadores, dado que estos gemelos al ser monocigóticos tendrían igual carga genética, y lleva a proponer que todas la variantes fenotípicas son secundarias a la misma alteración genética y a un grado variable de expresión en la función de PAX3.
Se ha propuesto que el desarrollo de la región frontonasal es sensible a la actividad del factor de transcripción codificado por el gen PAX3, pues los defectos en esa región ocurren en todos los afectados con mutaciones en este gen, explicando así cómo la totalidad de casos de SW tipo 1 tienen distopia cantorum (16) —definida cualitativamente como el desplazamiento lateral del canto ocular interno y cuantitativamente como un IW mayor de 1.95 (17)—; de igual forma, los defectos pigmentarios y el grado de pérdida de la audición varía entre los individuos afectados, como ocurrió en la familia aquí reportada donde las alteraciones del iris, la poliosis y la hipoacusia tuvieron expresividad variable, mientras que la distopia cantorum estuvo presente en la totalidad de los casos.
Los genes de la familia PAX también participan en el desarrollo de las extremidades. El SW tipo 3, que también presenta distopia cantorum acompañada de alteraciones musculo-esqueléticas producto de mutaciones en el gen PAX3, se manifiesta cuando las mutaciones en dicho gen son heredadas en homocigosis (5).
El SW se puede diagnosticar y clasificar clínicamente utilizando los criterios establecidos por el Consorcio Internacional para el Estudio del Síndrome de Waardenburg (18). El SW tipo 1 se caracteriza, entre otros hallazgos clínicos, por presentar distopia cantorum, mientras que en el tipo 2 está ausente (Tablas 1 y 2) (8); el tipo 3, también llamado síndrome de Klein Waardenburg, además de la distopia cantorum, se asocia con alteraciones músculo-esqueléticas en miembros superiores y el tipo 4 o síndrome de Shah-Waardenburg presenta como característica distintiva una asociación con la enfermedad de Hirschsprung (19).
Tabla 2. Criterios diagnósticos según el Consorcio Internacional del síndrome de Waardenburg tipo 1 y 2.
|
Criterios mayores |
G1 |
G2 |
M |
A |
|
Pérdida de la audición sensorial-neural congénita |
|
|
X |
|
|
Alteraciones en la pigmentación del iris |
X |
X |
X |
X |
|
Hipopigmentación del pelo (poliosis) |
|
|
X |
X |
|
Distopia cantorum |
X |
X |
X |
X |
|
Criterios menores |
G1 |
G2 |
M |
A |
|
Leucoderma congénita |
|
|
|
|
|
Raíz nasal ancha/prominente |
X |
X |
X |
X |
|
Hipoplasia de las alas nasales |
|
|
|
|
|
Encanecimiento prematuro |
|
|
|
X |
G1: Gemelo 1; G2: Gemelo 2; M: Madre; A: Abuela. Fuente: Elaboración con base en Krishtul et al. (24).
Dadas las diferencias fenotípicas y patrones de herencia específicos en los diferentes tipos de SW (Tabla 1 y 2), se puede hacer un diagnóstico diferencial en ausencia de pruebas moleculares. El diagnóstico del SW tipo 1 se realiza cuando están presentes dos criterios mayores más un criterio menor o un criterio mayor más dos menores. El SW tipo 2 se diagnostica cuando están presentes dos criterios mayores y hay ausencia de distopia cantorum.
En el heredograma de la familia aquí reportada se encontró un patrón de herencia autosómico dominante de las características clásicas de SW y se descartaron los tipos 3 y 4 teniendo en cuenta la ausencia de hipoplasia, sindactilia, contracturas u otras alteraciones musculo esqueléticas en miembros superiores y de signos y síntomas sugestivos de enfermedad de Hirschprung. Dada la presencia de distopia cantorum en todos los casos descritos, se descartó el tipo 2 y se hizo diagnóstico de SW tipo 1 en la familia.
Además, otro hallazgo relevante en el heredograma fue la presencia de fisuras orofaciales en los gemelos y un familiar por línea paterna en IV grado de consanguinidad (Figura 2; individuo III-1). Si bien esta alteración ha sido descrita en el SW (13), es una anomalía poco observada que fue reportada en población colombiana con una frecuencia de 3.6% (4) y que sugiere que el fenotipo del SW podría ser influenciado por genes modificadores, en este caso alelos de origen paterno (18).
Además, en los gemelos y en su abuela se encontró estrabismo convergente, característica no descrita para el SW (26) que podría también ser explicada por genes modificadores del fenotipo, dado que no estaba presente en todas las generaciones de afectados y sí en los gemelos. Sin embargo, el estrabismo también podría estar relacionado en los gemelos por el consumo de alcohol de la madre en el primer trimestre del embarazo, teniendo en cuenta que los dos presentan talla y peso menor de 2 desviaciones estándar, características tampoco incluidas en el fenotipo típico del SW pero que sí son observadas en el síndrome alcohólico fetal (20,21).
En la búsqueda realizada en las bases de datos médicas, solo se encontraron dos publicaciones de gemelos monocigóticos con SW, ninguna de ellas después del año 2000 (22,23). Esto mostró que si bien el SW es una enfermedad poco frecuente —1 por 40000 habitantes—, su diagnóstico en gemelos monocigóticos como los aquí descritos es aún más raro.
Los hallazgos aquí reportados muestran la variabilidad de las manifestaciones fenotípicas del SW tipo 1 dentro de una familia y especialmente en gemelos monocigóticos, lo que se ha explicado por la expresión variable de los genes específicos o la interacción de ellos con genes modificadores. Dada la exposición de los gemelos en el primer trimestre a alcohol, se propone que la expresión variable del SW sería influenciada por exposiciones a agentes medioambientales.
Conflicto de intereses
Ninguno declarado por los autores
Financiación
Ninguna declarada por los autores
Agradecimientos
A todos los integrantes de la familia aquí reportada.
Referencias
1. Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary of the iris and head hair and with congenital deafness. Am. J. Hum. Genet. 1951;3(3):195-253.
2. Read AP, Newton VE. Waardenburg syndrome. J. Med. Genet. 1997;34(8):656-65. http://doi.org/bn4pbc.
3. Nayak CS, Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann. Otol. Rhinol Laryngol. 2003;112(9):817-20. http://doi.org/bds5.
4. Tamayo ML, Gelvez N, Rodríguez M, Flórez S, Varon C, Medina D, et al. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Am. J. Med. Genet. A. 2008;46A(8):1026-31. http://doi.org/b2qx68.
5. Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int. J. Dermatol. 1999;38(9):656-63. http://doi.org/c6b72k.
6. Reynolds JE, Meyer JM, Landa B, Stevens CA, Arnos KS, Israel J, et al. Analysis of variability of clinical manifestations in Waardenburg syndrome. Am. J. Med. Genet. 1995;57(4):540-7. http://doi.org/bp2vzj.
7. Soni CR, Kumar G. Child neurology: a patient with dissimilar eye color and deafness. Neurology. 2010;74(8):e25-6. http://doi.org/fs9ww5.
8. Charrow J. Different color eyes. Waardenburg syndrome. Pediatr. Ann. 2007;36(5):277-8. http://doi.org/bds6.
9. Mallory SB, Wiener E, Nordlund JJ. Waardenburg's syndrome with Hirschsprung's disease: a neural crest defect. Pediatr. Dermatol. 1986;3(2):119-24. http://doi.org/d7nxw3.
10. Mehta M, Sethi S, Pushker N, Bajaj MS, Ghose S. Delayed presentation of children with Waardenburg Syndrome. J. Pediatr. Ophthalmol Strabismus. 2010;47(6):382-3. http://doi.org/dj62mq.
11. Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschprung disease (Shah-Waardenburg Syndrome). Nat. Genet. 1996;12(4):442-4. http://doi.org/fr2cxt.
12. Jang MA, Lee T, Lee J, Cho EH, Ki -S. Identification of a Novel De Novo Variant in the PAX3 Gene in Waardenburg Syndrome by Diagnostic Exome Sequencing: The First Molecular Diagnosis in Korea. Ann. Lab. Med. 2015;35(3):362-5. http://doi.org/bds9.
13. Eigelshoven S, Kameda G, Kortüm AK, Hübsch S, Angerstein W, Singh P, et al. Waanderburg syndrome type I with heterochromia iridis and circumscribed hypopigmentation of the skin. Pediatr. Dermatol. 2009;26(6):759-61. http://doi.org/c2tdft.
14. Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 2008;21(6):627-45. http://doi.org/ftsrsg.
15. Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010;31(4):391-406. http://doi.org/btppw2.
16. Milunsky JM. Waardenburg Syndrome Type I. In: Pagon RA, Adam MP, Ardinger HH, Wallac SE, Amemiya A, Bean L, et al., editors. Gene Reviews. Seattle: University of Washington; 1993-2001.
17. Demirci GT, Atis G, Altunary IK. Waardenburg Syndrome type 1: A case report. Dermatol Online J. 2011;17(11):3.
18. Online Mendelian Inheritance in Man®. Entry 193500 Waardenburg syndrome, type 1; WS1. Baltimore: Johns Hopkins University;1986 [updated 2010 Aug 3; cited 2016 Mar 30]. Available from: http://goo.gl/fzPzKx.
19. Tagra S, Talwar AK, Walia RL, Sidhu P. Waardenburg syndrome. Indian J. Dermatol. Venerol. Lepprol. 2006;72(4):326.
20. Zozaya-Aldana B, Medina-Rodríguez I. Alteraciones oculares en el síndrome alcohólico fetal. Rev. Cubana Obstet. Ginecol. 2011;30(1):100-9.
21. Jones KL, Jones MC, del Campo M. Smith’s Recognizable Patterns of Human Malformation. 7th ed. Philadelphia: Elsevier Saunders; 2013.
22. Carakushansky G, Berthier C. [Waardenburg's syndrome. Clinical and genetical study of a family with affected twins]. AMB Rev Assoc Med Bras. 1975;21(5):159-60. Portuguese.
23. Suyugül Z, Tüysüz B, Tükenmez F, Başaran M, Cenani A. Waardenburg syndrome: variable phenotypic expression in monozygotic twins. Clin. Dysmorphol. 1998;7(1):77-8.
http://doi.org/dgqfvc.
24. Krishtul A, Galadari I. Waardenburg syndrome: Case report. Int. J. Dermatol. 2003;42(8):651-2. http://doi.org/ddmqxn.
25. Wang J, Li S, Xiao X, Wang P, Guo X, Zhang Q. PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1. Mol. Vis. 2010;16:1146–53.
26. Nasser-Paranaíba LM, Ribeir-Paranaíba LM, Frota AC, Gomes A, Versiani G, Martelli-Júnior H. Síndrome de Waardenburg - aspectos oftalmológicos e critérios de diagnóstico: relatos de casos. Arq. Bras. Oftalmol. 2012;75(5):352-5. http://doi.org/bds8.
Ingrid Yoryeth Bastidas Pedreros
“Ejercicios de ilustración anatómica”
Universidad Nacional de Colombia
Referencias
Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary of the iris and head hair and with congenital deafness. Am. J. Hum. Genet. 1951;3(3):195-253.
Read AP, Newton VE. Waardenburg syndrome. J. Med. Genet. 1997;34(8):656-65. http://doi.org/bn4pbc.
Nayak CS, Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann. Otol. Rhinol Laryngol. 2003;112(9):817-20. http://doi.org/bds5.
Tamayo ML, Gelvez N, Rodríguez M, Flórez S, Varon C, Medina D, et al. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Am. J. Med. Genet. A. 2008;46A(8):1026-31. http://doi.org/b2qx68.
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int. J. Dermatol. 1999;38(9):656-63. http://doi.org/c6b72k.
Reynolds JE, Meyer JM, Landa B, Stevens CA, Arnos KS, Israel J, et al. Analysis of variability of clinical manifestations in Waardenburg syndrome. Am. J. Med. Genet. 1995;57(4):540-7. http://doi.org/bp2vzj.
Soni CR, Kumar G. Child neurology: a patient with dissimilar eye color and deafness. Neurology. 2010;74(8):e25-6. http://doi.org/fs9ww5.
Charrow J. Different color eyes. Waardenburg syndrome. Pediatr. Ann. 2007;36(5):277-8. http://doi.org/bds6.
Mallory SB, Wiener E, Nordlund JJ. Waardenburg's syndrome with Hirschsprung's disease: a neural crest defect. Pediatr. Dermatol. 1986;3(2):119-24. http://doi.org/d7nxw3.
Mehta M, Sethi S, Pushker N, Bajaj MS, Ghose S. Delayed presentation of children with Waardenburg Syndrome. J. Pediatr. Ophthalmol Strabismus. 2010;47(6):382-3. http://doi.org/dj62mq.
Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschprung disease (Shah-Waardenburg Syndrome). Nat. Genet. 1996;12(4):442-4. http://doi.org/fr2cxt.
Jang MA, Lee T, Lee J, Cho EH, Ki -S. Identification of a Novel De Novo Variant in the PAX3 Gene in Waardenburg Syndrome by Diagnostic Exome Sequencing: The First Molecular Diagnosis in Korea. Ann. Lab. Med. 2015;35(3):362-5. http://doi.org/bds9.
Eigelshoven S, Kameda G, Kortüm AK, Hübsch S, Angerstein W, Singh P, et al. Waanderburg syndrome type I with heterochromia iridis and circumscribed hypopigmentation of the skin. Pediatr. Dermatol. 2009;26(6):759-61. http://doi.org/c2tdft.
Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 2008;21(6):627-45. http://doi.org/ftsrsg.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010;31(4):391-406. http://doi.org/btppw2.
Milunsky JM. Waardenburg Syndrome Type I. In: Pagon RA, Adam MP, Ardinger HH, Wallac SE, Amemiya A, Bean L, et al., editors. Gene Reviews. Seattle: University of Washington; 1993-2001.
Demirci GT, Atis G, Altunary IK. Waardenburg Syndrome type 1: A case report. Dermatol Online J. 2011;17(11):3.
Online Mendelian Inheritance in Man®. Entry 193500 Waardenburg syndrome, type 1; WS1. Baltimore: Johns Hopkins University;1986 [updated 2010 Aug 3; cited 2016 Mar 30]. Available from: http://goo.gl/fzPzKx.
Tagra S, Talwar AK, Walia RL, Sidhu P. Waardenburg syndrome. Indian J. Dermatol. Venerol. Lepprol. 2006;72(4):326.
Zozaya-Aldana B, Medina-Rodríguez I. Alteraciones oculares en el síndrome alcohólico fetal. Rev. Cubana Obstet. Ginecol. 2011;30(1):100-9.
Jones KL, Jones MC, del Campo M. Smith's Recognizable Patterns of Human Malformation. 7th ed. Philadelphia: Elsevier Saunders; 2013.
Carakushansky G, Berthier C. [Waardenburg's syndrome. Clinical and genetical study of a family with affected twins]. AMB Rev Assoc Med Bras. 1975;21(5):159-60. Portuguese.
Suyugül Z, Tüysüz B, Tükenmez F, Başaran M, Cenani A. Waardenburg syndrome: variable phenotypic expression in monozygotic twins. Clin. Dysmorphol. 1998;7(1):77-8. http://doi.org/dgqfvc.
Krishtul A, Galadari I. Waardenburg syndrome: Case report. Int. J. Dermatol. 2003;42(8):651-2. http://doi.org/ddmqxn.
Wang J, Li S, Xiao X, Wang P, Guo X, Zhang Q. PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1. Mol. Vis. 2010;16:1146-53.
Nasser-Paranaíba LM, Ribeir-Paranaíba LM, Frota AC, Gomes A, Versiani G, Martelli-Júnior H. Síndrome de Waardenburg - aspectos oftalmológicos e critérios de diagnóstico: relatos de casos. Arq. Bras. Oftalmol. 2012;75(5):352-5. http://doi.org/bds8.
Cómo citar
APA
ACM
ACS
ABNT
Chicago
Harvard
IEEE
MLA
Turabian
Vancouver
Descargar cita
Licencia
Derechos de autor 2016 Revista de la Facultad de Medicina

Esta obra está bajo una licencia Creative Commons Reconocimiento 3.0 Unported.
-


















