



Publicado
A DFT study on Dichloro{(E)-4-dimethylamino-N′-[(pyridin-2 yl)methylidene-κN]benzohydrazide-κO}M2+ (M=Zn, Cu, Ni and Co) complexes: Effect of the metal over association energy and complex geometry
Un estudio DFT en complejos Dicloro {(E) -4-dimetilamino-N '- [(piridin-2 il) metilideno-κN] benzohydrazide-κO} M2 + (M = Zn, Cu, Ni, Fe, Mn, Ca y Co): efecto del metal sobre la energía de asociación y de geometría del complejo
DOI:
https://doi.org/10.15446/rev.colomb.quim.v45n3.57351Palabras clave:
Hydrazones, Binding energies, Single-Point, DFT, complexes (en)hidrazona, asociación de energía, SPE, DFT, complejos (es)
Descargas
The molecular geometry of (E)-4-dimethylamino-N′-[(pyridin-2-yl)methylidene- N]benzohydrazide (C15H16N4O) complexed with M2+ (M=Zn, Cu, Ni, Fe, Mn, Ca and Co) ions were calculated, using density functional theory (B3LYP) with 6-31G(d, p) basis set. Vibrational frequencies were computed in order to verify the absence of imaginary vibrational frequencies, fact that confirms the global minimum in geometry optimization. Molecular geometry parameters (bond lengths and angles) for Cu2+ and Zn2+ complexes were compared with crystallographic data previously reported, showing good correlation. Binding energies for all complexes were computed at B3LYP/6-31G++(d, p) level of theory. These calculations indicate that Cu-L is the lowest favorable complex, Cu2+ corresponds to the smallest cation on the present study. In the other hand, Ca-L, one of the less favorable complex, corresponds to the biggest cation analyzed in the present study. Molecular orbital analysis was carried out showing variations in energy differences between HOMO-LUMO values in function of the metallic ion employed.
La geometría molecular de la (E)-4-dimetilamino-N′-[(piridin-2-il)metilideno- N]benzohidrazida (C15H16N4O) acomplejada con iones M2+ (M=Zn, Cu, Ni, Fe, Mn, Ca y Co) se calculó usando la teoría funcional de densidad (B3LYP) empleando un conjunto de bases 6-31G(d, p). Las frecuencias vibracionales fueron calculadas con el propósito de comprobar la ausencia de frecuencias vibracionales imaginarias, hecho que confirma el mínimo global en la optimización de la geometría. Los parámetros de la geometría molecular (longitudes de enlace y ángulos) para los complejos de Cu2+ y Zn2+ fueron comparados con datos cristalográficos previamente reportados, mostrando una buena correlación. Las energías de asociación para todos los complejos fueron determinadas a un nivel de teoría B3LYP/6-31G++(d, p) mostrando que el complejo menos favorable es Cu-L, correspondiente al catión más pequeño del estudio. Por otro lado Ca-L, uno de los menos estables, corresponde al catión más grande analizado. Se llevó a cabo un análisis de orbitales moleculares en el cual los complejos exhibieron diferentes valores de diferencia de energía HOMO-LUMO en función del metal empleado.
Doi: https://doi.org/10.15446/rev.colomb.quim.v45n3.57351
A DFT study on Dichloro {(E)-4-dimethylamino-N'-[(pyridin-2-yl) methylideneKN] benzohydrazide-k0}M2+ (M = Zn, Cu, Ni, Fe, Mn, Ca and Co) complexes: Effect of the metal over association energy and complex geometry
Un estudio DFT en complejos Dicloro {(E)-4-dimetilamino-N'-[(piridin-2-il) metilideno-KN] benzohidrazida-K0} M2+ (M = Zn, Cu, Ni, Fe, Mn, Ca y Co): efecto del metal sobre la energía de asociación y la geometría del complejo
Um estudo DFT nos complexos Dicloro {(E)-4-dimetilamino-N'-[(piridin-2-il) metilideno -KN] benzohidrazida-K0} M2+ (M = Zn, Cu, Ni, Fe, Mn, Ca y Co): efeito do metal sobre a energia de associação e a geometria do complexo
Gustavo Gutiérrez1, Mónica A. Gordillo2, Manuel N. Chaur2,*
1 Departamento de Ciencias Farmacéuticas, Universidad Icesi, Cali-Colombia
2 Departamento de Química, Universidad del Valle, A.A., 25360 Cali-Colombia.
* Autor para correspondencia: manuel.chaur@correounivalle.edu.co
Article citation:
Gutiérrez, G.; Gordillo, M. A.; Chaur, M. N. A DFT study on Dichloro {(E)-4-dimethylamino-N'-[(pyridin-2-yl) methylidene-KN] benzohydrazide-KO}M2+ (M = Zn, Cu, Ni, Fe, Mn, Ca and Co) complexes: Effect of the metal over association energy and complex geometry Rev. Colomb. Quim. 2016, 45 (3), 28-32. DOI: https://doi.org/10.15446/rev.colomb.quim.v45n3.57351.
Recibido: 10 de Mayo de 2016. Aceptado: 8 de Septiembre de 2016.
Abstract
The molecular geometry of (E)-4-dimethylamino-N'-[(pyridin-2yl)methylidene-kN]benzohydrazide (C15H16N4O) complexed with M2+ (M=Zn, Cu, Ni, Fe, Mn, Ca and Co) ions was calculated, using density functional theory (B3LYP) with 6-31G(d, p) basis set. Vibrational frequencies were computed in order to verify the absence of imaginary vibrational frequencies, fact that confirms the global minimum in geometry optimization. Molecular geometry parameters (bond lengths and angles) for Cu2+ and Zn2+ complexes were compared with crystallographic data previously reported, showing good correlation. Binding energies for all complexes were computed at the B3LYP/6-31G++(d, p) level of theory. These calculations indicate that Cu-L is the lowest favorable complex, Cu2+ corresponds to the smallest cation on the present study. On the other hand, Ca-L, one of the less favorable complex, corresponds to the largest cation analyzed in the present study. Molecular orbital analysis was carried out showing variations in ΔE HOMO-LUMO values as a function of the metallic ion employed.
Keywords: Hydrazones, Binding energies, Single-Point, DFT, complexes.
Resumen
La geometría molecular de la (E)-4-dimetilamino-N'-[(piridin-2-il) metilideno-kN] benzohidrazida (C15H16N4O) acomplejada con iones M2+ (M=Zn, Cu, Ni, Fe, Mn, Ca y Co) se calculó usando la teoría funcional de densidad (B3LYP) empleando un conjunto de bases 6-31G(d, p). Las frecuencias vibracionales fueron calculadas con el propósito de comprobar la ausencia de frecuencias vibracionales imaginarias, hecho que confirma el mínimo global en la optimización de la geometría. Los parámetros de la geometría molecular (longitudes de enlace y ángulos) para los complejos de Cu2+ y Zn2+ fueron comparados con datos cristalográficos previamente reportados, mostrando una buena correlación. Las energías de asociación para todos los complejos fueron determinadas a un nivel de teoría B3LYP/6-31G++(d, p) mostrando que el complejo menos favorable es Cu-L, correspondiente al catión más pequeño del estudio. Por otro lado Ca-L, uno de los menos estables, corresponde al catión más grande analizado. Se llevó a cabo un análisis de orbitales moleculares en el cual los complejos exhibieron diferentes valores de ΔE HOMO-LUMO en función del metal empleado.
Palabras clave: hidrazona, energía de asociación, SPE, DFT, complejos.
Resumo
A geometria molecular da (E)-4-dimetilamino-N'-[(piridin-2-il) metilideno-kN] benzohidrazida (C15H16N4O) acomplexada com íons M2+ (M=Zn, Cu, Ni, Fe, Mn, Ca y Co) foi calculada usando a teoria funcional da densidade (B3LYP) utilizando um conjunto de bases 6-31G(d, p). As frequências vibracionais foram calculadas com o objetivo de comprovar a ausência de frequências vibracionais imaginárias, fato que confirma o mínimo global na otimização da geometria. Os parâmetros da geometria molecular (longitudes de enlace e ângulos) para os complexos de Cu2+ y Zn2+ foram comparados com dados cristalográficos previamente reportados e mostraram boa correlação. As energias de associação para todos os complexos foram determinadas ao nível de teoria B3LYP/6-31G++(d, p) mostrando que o complexo menos favorável é Cu-L, correspondente ao cátion mais pequeno do estudo. Por outro lado Ca-L, um dos menos estáveis, corresponde ao cátion mais grande analisado. Foi feita uma análise de orbitais moleculares no qual os complexos exibiram diferentes valores de ΔE HOMO-LUMO em função do metal utilizado.
Palavras-Chave: hidrazona, associação de energia, SPE, DTF, complexos.
Introduction
Calculations
Hydrazones are compounds with interesting properties in various research fields such as pharmacology (1-3) antibiotics (4), analytical purposes (J) among others (6-7). In this particular case, the interest is specially focused on the multiple hydrazones dynamics that are important features to the configurational dynamics (imine bond) and the ability to coordinate (terpyridine-like). These characteristics confer applicability in the development of molecular machines and systems for information storage (8-9). Pyridine-2-carboxaldehyde, aroyl or acyl hydrazone derivatives exhibit thermal or photo-induced reversible E/Z isomerization where the Z-isomer is in a thermodynamic metastable state stabilized by an intramolecular hydrogen bond (10-12). Additionally, the terpyridine-like framework of most hydrazones allows them to coordinate metal centers (9, 13). Induced isomerization and metal coordination constitute configurational changes in the short term. Reversible chemical identity modifications are available as well and represent constitutional dynamics, which are especially useful for long-term information storage applications (14).
Density Functional Theory (DFT) methods have been widely used for calculating molecular optimized geometries and spectral properties, for instance NMR (1J), UV (16), IR (17) and Raman (18), with good correlations with those obtained experimentally. In this sense, one of the most widely used method in computational chemistry is the Becke's three-parameter hybrid (19) and Lee, Yang and Parr correlation functional (B3LYP ) (20). This method has proven to be an especially useful approach in the computational study of inorganic (21) and organometallic complexes (22), as well as organic compounds. For instance, in a previous work it was found good correlation between the experimental data of a 2-pyridinecarboxaldehyde derivative and its computed results at two levels of theory (12).
Typically, DFT studies may or may not use diffuse s and p functions for non-hydrogen (6-31G+) atoms and also hydrogen atoms (6-31G++), but the use of d-polarization functions at least for non-hydrogen atoms (6-31G (d)) (23) in organic systems seems mandatory in order to obtain reasonable accuracy; the use of basis that employ polarization functions for all atoms (6-31G (d, p)) is common (24, 2J).
Furthermore, Sangeetha et al. (26) reported the crystal structure and other spectral properties of (E)-4-dimethylamino-N'-[(pyridin-2-yl)methylidene]benzohydrazide copper (II) complex. Previously, the crystal structure of the same ligand participating in a Zn (II) complex was reported by this research group (27). Nevertheless, as far as it is known, there are no theoretical studies for the titled complexes reported in literature. In that way, in order to understand the effect of metallic ion over complex properties, the full-optimized geometric parameters of the titled ligand (C15H16N4O) uncomplexed and associated with four transition metallic ions in the ground state, association energies, and Highest Occupied- and Lowest Unoccupied Molecular Orbitals (HOMO-LUMO) energy gaps were calculated. In this order of ideas, improving the comprehension about complex formation may lead to the development of coordination brakes capable of performing a controlled movement or result in the rational use of certain metal ions in dynamic combinatorial chemistry. Therefore, these results may show the possible use of this kind of complexes in supramolecular structures with specific applications among them in metalo-supramolecular chemistry, coordination polymers based on hydrazones and molecular machines.
Calculations
Molecular geometries of the free ligand and its M2+ (M = Zn, Cu, Ni, Fe, Mn, Ca and Co) complexes in the ground state were optimized by DFT:B3LYP method with 6-31G (d, p) basis set. Theoretical IR spectra were computed at the same level of theory to confirm the absence of imaginary frequencies, and single-point energy calculations were performed using B3LYP/6-31G++ (d, p) employing Gauss View 5 as a graphic interface (28) and Gaussian 09W64 (29) for running the calculations. Binding energies were calculated as described by Lee (30) using equation [1].

where Ecomplex is the single-point energy (SPE) of the complex, EM2+ is the computed SPE for the chloride salt of each metal and EC15H16N4O is the ligand SPE.
Results and discussion
The crystal morphology of 2-pyridine-carboxaldehyde-4-dimethylaminobenzoylhydrazone is monoclinic with space group P21/c and it is presented as an ethanolic solvate with cell dimensions a = 6.670 (4) Å, b = 29.778 (3) Å, c = 8.010 (2) Å and V = 1575.96 Å3 (31). The optimized geometry was calculated at the DFT: B3LYP / 6-31G (d, p) level of theory with a good theoretical-experimental correlation, the relative error was lower than 2.7% for both, bond lengths and angles.
On the one hand, the crystal structure of the copper complex is monoclinic with an space group C/2c and cell dimensions a = 19.849 (3) Å, b = 17.675 (2) Å, c = 11.876 (2) Å and V = 3386.91 Å3 (26). Likewise, the zinc complex exhibits a monoclinic geometric structure with an space group P21/c and cell dimensions a = 19.849 (3) Å, b = 13.586 (5) Å, c = 7.598 (9) Å and V = 1670.36 Å3 (27). Since there are no crystallographic reports for the aforementioned ligand complexed with more metals than copper and zinc, theoretical-experimental correlation is only possible for the mentioned metals; correlation graphs of selected geometrical parameters are shown in Figure 1.
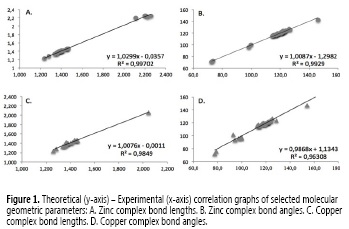
Accordingly, linear regression is an useful tool in comparative studies of two set of data where the slope is a proportion between response variation and input variables, thus, a perfect correlation would have a slope equal to one. Higher or lower values represent over or underestimation of the calculated values regarding the experimental data, respectively. The intercept is, by definition, the value of the response variable when the input is equal to zero, hence, negative or positive intercept values represent quantitatively the over- or underestimation of calculations.
Finally, R2 values provide the percentage of response variability that is explained by the variation of the independent variable, thus, the difference between R2 value and one represents an indirect measure of random error (32). In that way, the calculations in this research are slightly overestimated for bond angles and bond lengths in the zinc complex and underestimated in the copper complex. The latter showed the higher random error.
The biggest discrepancies for both complexes are related with the angle and length of the chlorine-metal bond. This error is explained by the high influence of intermolecular interactions in solid phase, neglected in these computational calculations, which assume an isolated molecule in the gas phase; N-H-Cl along [001] direction stacking.
Therefore, we excluded the aforementioned bonds in error rate calculations. On the one hand, copper complex showed acceptable average errors of 1.12 and 1.46% for bond lengths and angles respectively. On the other hand, zinc complex report 0.83 and 0.66% for bond lengths and angles, respectively.
Results from SPE calculations were performed at DFT: B3LYP/6-31G++(d, p) level of theory for the full-optimized geometries of the titled ligand, the ligand complexed with zinc, copper, nickel, manganese, iron, calcium and cobalt, and the chloride of each metal without restrictions, as well as metallic center geometry parameters are presented below in Table 1. The optimized structure of each complex along with the ORTEP representation of the zinc complex's structure is shown in Figure 2.


Association energy results showed that the most favorable complex formation is the one formed between the ligand and manganese (II) (-74.8 kcal/mol) while the less favorable complexes are those formed with Copper and Zinc 2+ ions (-25.8 and -26.2 kcal/mol, respectively). Noteworthily, figures 2A and 2B show that although Cu2+ and Zn2+ ions are capable of fitting in the N-N-O pocket of the ligand, the small size of these ions results in longer distances between the metal cations and atoms, from the ligand, participating in the coordination framework.
Ca2+ is the largest ion under the present study and, as observed in figure 2C, is too voluminous fit in the N-N-O pocket inducing again longer distances, decreasing the efectiveness of interaction and therefore the complex stability in both cases. Nickel (II) complex showed a particular behavior since exhibits the less N1-N2-O1 angle while N1-MO1 angle and the N2-M, O1-M and N2-M distances are comparable to other complexes (Co, Fe or Mn, i. e.) indicating an especial structural modification of the ligand closing the N1-O distance, probably to promoting the interaction ligand-metal.
Since HOMO-LUMO is mainly a mathematical model that represents electronic density around atoms and not directly experimentally observable parameters, usually they are physically explained as the ionization potential and electron affinity, respectively. However, HOMO-LUMO energy gap and other interactions between both molecular orbitals studied in Frontier Molecular Orbitals Theory (FMOT) are important for chemical reactivity of molecular systems. As observed in figure 3 the ligand and the zinc complex HOMO-FUMO transitions are characterized by a subtle electronic displacement from the phenyl-hydrazone moiety to the pyridine ring. In contrast, other complexes have the HOMO localized at the metallic center and their HOMO-LUMO transitions are characterized by an electronic displacement towards the phenyl-hydrazone and pyridine ring moieties, this is specially observed for the copper complex.

Conclusions
Optimized geometries of the titled ligand and their respective complexes of seven metallic 2+ ions (Zn, Cu, Ni, Mn, Fe, Ca and Co) were calculated and compared with crystallographic reports for the ligand, zinc and copper complexes with good theoretical-experimental correlation.
The main discrepancy between crystal and calculated structures was found in the chloride-metal-chloride geometry due to computed geometries that were carry out in gas phase for an isolated molecule, approach that neglects intermolecular interactions observed in the solid state structure (N-H ... Cl along [001] direction stacking, i. e.). Association energy calculations showed that the most favorable complex formation occurs in presence of Mn2+ while Zn2+, Ca2+ and Cu2+ are the less favorable complexes.
Acknowledgments
We greatly thank to Vicerrectoría de Investigaciones and Centro de Excelencia en Nuevos Materiales (CENM) from Universidad del Valle for their generous financial support.
References
1. Giziroglu, E.; Aygün, M.; Sarikurkcu, C.; Kazar, D.; Orhan, N.; Firinki, H. et al. Synthesis, characterization and antioxidant activity of new dibasic tridentate ligands: X-ray crystal structures of DMSO adducts of I, 3-dimethyl-5-acetylbarbituric acid o-hydroxybenzoyl hydrazone copper(II) complex Inorg. Chem. Commun. 2013, 36, 199-205. DOI: https://doi.org/10.1016/j.inoche.2013.09.013.
2. Kovács, D.; Wölfling, J.; Szabó, N.; Szécsi, M.; Minorics, R.; Zupkó, I. et al. Efficient access to novel androsteno-17-(1',3',4')-oxadiazoles and 17β-(1',3',4')-thiadiazoles via N-substituted hydrazone and N,N'-disubstituted hydrazine intermediates, and their pharmacological evaluation in vitro Eur. J. Med. Chem. 2015, 98, 13-29. DOI: https://doi.org/10.1016/j.ejmech.2015.05.010.
3. S. Rollas, S. Küçükgüzel, Molecules, 12, 1910 (2007).
4. Sridhar, S.K.; Saravanan, M.; Ramesh, A. Synthesis and antibacterial screening of hydrazones, Schiff and Mannich bases of isatin derivatives. Eur. J. Med. Chem., 2001, 36 (7), 615-625. DOI: https://doi.org/10.1016/s0223-5234(01)01255-7.
5. Suvarapu, L.N.; Seo, Y.K.; Baek, S. O.; Ammireddy, V.R. Review on analytical and biological applications of hydrazones and their metal complexes. E-J. Chem. (Online), 2012, 9 (3), 1288-130. DOI: https://doi.org/10.1155/2012/534617.
6. Su, X.; Aprahamian, I.; Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43 (6), 1963-1981. DOI: https://doi.org/10.1039/c3cs60385g.
7. Van Dijken, D. J.; Kovaricek, P. S.; Ihrig, P.; Hecht, S. Acylhydrazones as Widely Tunable Photoswitches. J. Am. Chem. Soc. 2015, 137 (47), 14982-14991. DOI: https://doi.org/10.1021/jacs.5b09519.
8. Chaur, M.N.; Collado, D.; Lehn, J. M. Configurational and constitutional information storage: multiple dynamics in systems based on pyridyl and acyl hydrazones Chem. Eur. J 2011, 17 (1), 248-258 DOI: https://doi.org/10.1002/chem.201002308.
9. Romero, E.; D'Vries, R.; Zuluaga, F; Chaur, M. Multiple dynamics of hydrazone based compounds. J. Braz. Chem. Soc. 2015, 26 (3) 1255-1273. DOI: https://doi.org/10.5935/0103-5053.20150092.
10. Landge, S.M.; Tkatchouk, E.; Benitez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. et al. Isomerization mechanism in hydrazone-based rotary switches: lateral shift, rotation, or tautomerization? J. Am. Chem. Soc. 2011, 133 (25), 9812-9823. DOI: https://doi.org/10.1021/ja200699v.
11. Chaur, M. N. Aroylhydrazones as potential systems for information storage: photoisomerization and metal complexation Rev. Colomb.Quim 2012, 41 (3), 349-358.
12. Gordillo, M.A.; Soto-Monsalve, M.; Gutiérrez, G; D'vries, R.; Chaur, M. N. Theoretical and experimental comparative study of a derivative from 2-pyridinecarboxaldehyde which exhibits configurational dynamics J. Mol. Struc. 2016, 1119, 286-295. DOI: https://doi.org/10.1016/j.molstruc.2016.04.055.
13. Fernández, M. A.; Barona, J. C.; Polo, D.; Chaur, M. N. Photochemical and electrochemical studies on lanthanide complexes of 6-(hydroxymethyl) pyridine-2-carboxaldehyde [2-methyl-pyrimidine-4, 6-diyl] bis-hydrazone. Rev. Colomb. Quim. 2014, 43 (1), 5-11.
14. Lehn, J. M. Conjecture: Imines as unidirectional photodriven molecular motors-motional and constitutional dynamic devices. Chem. Eur. J. 2006, 12 (23), 5910-5915. DOI: https://doi.org/10.1002/chem.200600489.
15. Bagno, A.; Saielli, G. Addressing the stereochemistry of complex organic molecules by density functional theory - NMR. WIREs Comput. Mol. Sci. 2015, 5 (2), 228-240. DOI: https://doi.org/10.1002/wcms.1214.
16. Preat, J.; Jacquemin, D.; Wathelet, V.; Fontaine, M.; Perpète, E.A. Thioindigo dyes: highly accurate visible spectra with TD-DFT. J. Phys. Chem. A, 2006, 128 (6), 2072-2083. DOI: https://doi.org/10.1021/ja056676h.
17. Wang, Y.; Liu, Q.; Qiu, L.; Wang, T.; Yuan, H. Lin, J. et al. Molecular structure, IR spectra, and chemical reactivity of cisplatin and transplatin: DFT studies, basis set effect and solvent effect. Spectrochim. Acta, Part A. 2015, 150, 902-908.
18. Sudha, S.; Sundaraganesan, N.; Kurt, M.; Cinar. M.; Karabacak, M. FT-IR and FTRaman spectra, vibrational assignments, NBO analysis and DFT calculations of 2-amino-4-chlorobenzonitrile. J. Mol. Struc. 2011, 985, 148. DOI: https://doi.org/10.1016/j.molstruc.2010.10.035.
19. Becke, A. D. Density - functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648-5652. DOI: https://doi.org/10.1063/1.464913.
20. Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785-789. DOI: https://doi.org/10.1103/physrevb.37.785.
21. Dudev, T.; Lim, C. Tetrahedral vs octahedral zinc complexes with ligands of biological interest: a DFT/CDM study. J. Am. Chem. Soc. 2000, 122 (45), 11146-11153. DOI: https://doi.org/10.1021/ja0010296.
22. Bagno, A.; Rastrelli, F; Saielli, G. Toward the complete prediction of the 1H and 13C NMR spectra of complex organic molecules by DFT methods: application to natural substances Chem. Eur. J. 2006, 12 (21), 5514-5525. DOI: https://doi.org/10.1002/chem.200501583.
23. Rablen, P. R.; Lockman, J. W.; Jorgensen, W. L. Ab initio calculations on hydrogen-bonded complexes of small organic molecules with water. J. Chem. Phys. A 1998, 102 (21), 3782-3797. DOI: https://doi.org/10.1021/jp980708o.
24. Szemik-Hojniak, A.; Oberda, K.; Deperasińska, I.; Nizhnik, Y. P.; Jerzykiewciz, L. A negligible CT character of the lowest excited state of a novel complex of zinc tetraphenylporphyrin with axially bonded 2-(4-methoxy-trans-styryl)quinoline-1-oxide ligand: Experimental studies and TD DFT/CAM B3LYP [6-31G(d,p)] calculations. Polyhedron 2015, 88, 190-198. DOI: https://doi.org/10.1016/j.poly.2014.12.025.
25. Maxwell, C. I.; Mosey, N. J.; Stan Brown, R. DFT Computational study of the methanolytic cleavage of DNA and RNA phosphodiester models promoted by the dinuclear Zn(II) complex of 1,3-Bis(1,5,9-triazacyclododec-1-yl)propane. J. Am. Chem. Soc. 2013, 135 (45), 17209-17222. DOI: https://doi.org/10.1021/ja4088264.
26. Sangeetha, N. R.; Pal, S.; Anson, C. E. Powell, A. K.; Pal, S. A one-dimensional assembly of copper(II) polyhedra via dual use of hydrogen-bonding and Π-Π interaction Inorg. Chem. Commun. 2000, 3 (8), 415-419. DOI: https://doi.org/10.1016/s1387-7003(00)00103-9.
27. Chaur, M. Dichlorido{(E)-4-dimethylamino-N'-[(pyridin-2-yl)methylidenejN] benzohydrazide-jO}zinc Acta Crystallogr. Sect. E: Struct. Rep. Online 2013, 69, m27. DOI: https://doi.org/10.1107/s1600536812049355.
28. GaussView, Version 5, Dennington, R.; Keith, T.; Millam, J. Semichem Inc., Shawnee Mission, KS, 2009.
29. Gaussian 09, Revision D.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H B et al , Gaussian, Inc , Wallingford CT, 2009.
30. Lee, G. Y. DFT studies of the zinc complexes of DNA bases Bull. Korean Chem. Soc. 2006, 27 (3), 419-422. DOI: https://doi.org/10.5012/bkcs.2006.27.3.419.
31 Sangeetha, N R ; Pal, S ; Pal S Copper(II)-activated transformation of azomethine to imidate: synthetic and structural studies. Polyhedron. 2000, 19 (28), 2713-2717. DOI: https://doi.org/10.1016/s0277-5387(00)00595-7.
32. Stockl, D.; Dewitte, K.; Thienpont L.M. Validity of linear regression in method comparison studies: is it limited by the statistical model or the quality of the analytical input data- Clin. Chem. 1998, 44 (11),2340-2346.
Referencias
Giziroglu, E.; Aygün, M.; Sarikurkcu, C.; Kazar, D.; Orhan, N.; Firinki, H. et al. Synthesis, characterization and antioxidant activity of new dibasic tridentate ligands: X-ray crystal structures of DMSO adducts of 1,3-dimethyl-5-acetyl-barbituric acid o-hydroxybenzoyl hydrazone copper(II) complex Inorg. Chem. Commun. 2013, 36, 199-205. DOI: https://doi.org/10.1016/j.inoche.2013.09.013
Kovács, D.; Wölfling, J.; Szabó, N.; Szécsi, M.; Minorics, R.; Zupkó, I. et al. Efficient access to novel androsteno-17-(1′,3′,4′)-oxadiazoles and 17β-(1′,3′,4′)-thiadiazoles via N-substituted hydrazone and N,N′-disubstituted hydrazine intermediates, and their pharmacological evaluation in vitro Eur. J. Med. Chem. 2015, 98, 13-29. DOI: http://dx.doi.org/10.1016/j.ejmech.2015.05.010
S. Rollas, S. Küçükgüzel, Molecules, 12, 1910 (2007).
Sridhar, S.K.; Saravanan, M.; Ramesh, A. Synthesis and antibacterial screening of hydrazones, Schiff and Mannich bases of isatin derivatives. Eur. J. Med. Chem., 2001, 36 (7), 615-625. DOI: http://dx.doi.org/10.1016/s0223-5234(01)01255-7
Suvarapu, L.N.; Seo, Y.K.; Baek, S. O.; Ammireddy, V.R. Review on analytical and biological applications of hydrazones and their metal complexes. E-J. Chem. (Online), 2012, 9 (3), 1288-130.DOI: https://doi.org/10.1155/2012/534617
Su, X.; Aprahamian, I.; Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43 (6), 1963-1981. DOI: https://doi.org/10.1039/c3cs60385g
van Dijken, D. J.; Kovaricek, P. S.; Ihrig, P.; Hecht, S. Acylhydrazones as Widely Tunable Photoswitches. J. Am. Chem. Soc. 2015, 137 (47), 14982-14991. DOI: https://doi.org/10.1021/jacs.5b09519
Chaur, M.N.; Collado, D.; Lehn, J. M. Configurational and constitutional information storage: multiple dynamics in systems based on pyridyl and acyl hydrazones. Chem. Eur. J. 2011, 17 (1), 248-258. DOI: http://dx.doi.org/10.1002/chem.201002308
Romero, E.; D’Vries, R.; Zuluaga, F.; Chaur, M. Multiple dynamics of hydrazone based compounds. J. Braz. Chem. Soc. 2015, 26 (3) 1255-1273. DOI: https://doi.org/10.5935/0103-5053.20150092
Landge, S.M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. et al. Isomerization mechanism in hydrazone-based rotary switches: lateral shift, rotation, or tautomerization? J. Am. Chem. Soc. 2011, 133 (25), 9812-9823. DOI: https://doi.org/10.1021/ja200699v
Chaur, M. N. Aroylhydrazones as potential systems for information storage: photoisomerization and metal complexation Rev. Colomb. Quim. 2012, 41 (3), 349-358
Gordillo, M.A.; Soto-Monsalve, M.; Gutiérrez, G.; D´vries, R.; Chaur, M. N. Theoretical and experimental comparative study of a derivative from 2-pyridinecarboxaldehyde which exhibits configurational dynamics J. Mol. Struc. 2016, 1119, 286-295. DOI: https://doi.org/10.1016/j.molstruc.2016.04.055
Fernández, M. A.; Barona, J. C.; Polo, D.; Chaur, M. N. Photochemical and electrochemical studies on lanthanide complexes of 6-(hydroxymethyl) pyridine-2-carboxaldehyde [2-methyl-pyrimidine-4, 6-diyl] bis-hydrazone. Rev. Colomb. Quim. 2014, 43 (1), 5-11.
Lehn, J. M. Conjecture: Imines as unidirectional photodriven molecular motors—motional and constitutional dynamic devices. Chem. Eur. J. 2006, 12 (23), 5910-5915. DOI: https://doi.org/10.1002/chem.200600489
Bagno, A.; Saielli, G. Addressing the stereochemistry of complex organic molecules by density functional theory‐NMR. WIREs Comput. Mol. Sci. 2015, 5 (2), 228-240. DOI: https://doi.org/10.1002/wcms.1214
Preat, J.; Jacquemin, D.; Wathelet, V.; Fontaine, M.; Perpète, E.A. Thioindigo dyes: highly accurate visible spectra with TD-DFT. J. Phys. Chem. A, 2006, 128 (6), 2072-2083. DOI: https://doi.org/10.1021/ja056676h
Wang, Y.; Liu, Q.; Qiu, L.; Wang, T.; Yuan, H. Lin, J. et al. Molecular structure, IR spectra, and chemical reactivity of cisplatin and transplatin: DFT studies, basis set effect and solvent effect. Spectrochim. Acta, Part A. 2015, 150, 902-908
Sudha, S.; Sundaraganesan, N.; Kurt, M.; Cinar. M.; Karabacak, M. FT-IR and FT-Raman spectra, vibrational assignments, NBO analysis and DFT calculations of 2-amino-4-chlorobenzonitrile. J. Mol. Struc. 2011, 985, 148. DOI: http://dx.doi.org/10.1016/j.molstruc.2010.10.035
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648-5652. DOI: http://dx.doi.org/10.1063/1.464913
Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785-789. DOI: http://dx.doi.org/10.1103/physrevb.37.785
Dudev, T.; Lim, C. Tetrahedral vs octahedral zinc complexes with ligands of biological interest: a DFT/CDM study. J. Am. Chem. Soc. 2000, 122 (45), 11146–11153. DOI: http://dx.doi.org/10.1021/ja0010296
Bagno, A.; Rastrelli, F.; Saielli, G. Toward the complete prediction of the 1H and 13C NMR spectra of complex organic molecules by DFT methods: application to natural substances. Chem. Eur. J. 2006, 12 (21), 5514-5525. DOI: https://doi.org/10.1002/chem.200501583
Rablen, P. R.; Lockman, J. W.; Jorgensen, W. L. Ab initio calculations on hydrogen-bonded complexes of small organic molecules with water. J. Chem. Phys. A 1998, 102 (21), 3782-3797. DOI: https://doi.org/10.1021/jp980708o
Szemik-Hojniak, A.; Oberda, K.; Deperasińska, I.; Nizhnik, Y. P.; Jerzykiewciz, L. A negligible CT character of the lowest excited state of a novel complex of zinc tetraphenylporphyrin with axially bonded 2-(4-methoxy-trans-styryl)quinoline-1-oxide ligand: Experimental studies and TD DFT/CAM B3LYP [6-31G(d,p)] calculations. Polyhedron 2015, 88, 190-198. DOI: http://dx.doi.org/10.1016/j.poly.2014.12.025
Maxwell, C. I.; Mosey, N. J.; Stan Brown, R. DFT Computational study of the methanolytic cleavage of DNA and RNA phosphodiester models promoted by the dinuclear Zn(II) complex of 1,3-Bis(1,5,9-triazacyclododec-1-yl)propane. J. Am. Chem. Soc. 2013, 135 (45), 17209-17222. DOI: https://doi.org/10.1021/ja4088264
Sangeetha, N. R.; Pal, S.; Anson, C. E. Powell, A. K.; Pal, S. A one-dimensional assembly of copper(II) polyhedra via dual use of hydrogen-bonding and π–π interaction Inorg. Chem. Commun. 2000, 3 (8), 415-419. DOI: http://dx.doi.org/10.1016/s1387-7003(00)00103-9
Chaur, M. Dichlorido{(E)-4-dimethylamino-N`-[(pyridin-2-yl)methylidene-jN]benzohydrazide-jO}zinc Acta Crystallogr. Sect. E: Struct. Rep. Online 2013, 69, m27. DOI: https://doi.org/10.1107/s1600536812049355
GaussView, Version 5, Dennington, R.; Keith, T.; Millam, J. Semichem Inc., Shawnee Mission, KS, 2009.
Gaussian 09, Revision D.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B. et al., Gaussian, Inc., Wallingford CT, 2009.
Lee, G. Y. DFT studies of the zinc complexes of DNA bases Bull. Korean Chem. Soc. 2006, 27 (3), 419-422.DOI: https://doi.org/10.5012/bkcs.2006.27.3.419
Sangeetha, N. R.; Pal, S.; Pal S. Copper(II)-activated transformation of azomethine to imidate: synthetic and structural studies. Polyhedron. 2000, 19 (28), 2713-2717. DOI: https://doi.org/10.1016/s0277-5387(00)00595-7
Stockl, D.; Dewitte, K.; Thienpont L.M. Validity of linear regression in method comparison studies: is it limited by the statistical model or the quality of the analytical input data? Clin. Chem. 1998, 44 (11), 2340-2346.
Cómo citar
IEEE
ACM
ACS
APA
ABNT
Chicago
Harvard
MLA
Turabian
Vancouver
Descargar cita
CrossRef Cited-by
1. J. Galindo Betancourth, N. E. Sánchez-Rodríguez, D. Giraldo-Dávila, M. Y. Combariza, M. N. Chaur. (2022). Self-Assembled Co(II) and Zn(II) Complexes with Soluble Bis(Hydrazone)Thiopyrimidine-Based Ligands: Electrochemical and Temperature-Dependent Properties. Russian Journal of Inorganic Chemistry, 67(14), p.2200. https://doi.org/10.1134/S0036023622600782.
2. Jungpil Kim. (2024). Investigating structural disparities in carbon nanoribbons and nanobelts through spectroscopies. Carbon Letters, 34(9), p.2447. https://doi.org/10.1007/s42823-024-00825-y.
3. Jungpil Kim. (2024). Unveiling Structural Variations in Armchair-Edge Coronoids by Spectroscopies. ACS Omega, 9(43), p.43956. https://doi.org/10.1021/acsomega.4c07966.
4. Manuel Noé Chaur Valencia, Elkin Libardo Romero, Gustavo Gutierrez, Mónica Soto Monsalve, Richard D´Vries, Héctor Fabio Zuluaga. (2018). Structural, spectroscopic, and theoretical analysis of a molecular system based on 2-((2-(4-chlorophenylhydrazone)methyl)quinolone. Revista Colombiana de Química, 47(2), p.63. https://doi.org/10.15446/rev.colomb.quim.v47n2.67115.
5. Harok Jeong, Sangmin Park, Junghoon Yang, Hye-Min Lee, Sangmin An, Yasuhiro Yamada, Jungpil Kim. (2022). Spectroscopic Distinction of Carbon Nanobelts and Nanohoops. SSRN Electronic Journal , https://doi.org/10.2139/ssrn.4191318.
6. Jungpil Kim. (2024). Spectroscopic Differentiation of Structural Transitions from Carbon Nanobelts to Carbon Nanotubes. The Journal of Physical Chemistry Letters, 15(44), p.11155. https://doi.org/10.1021/acs.jpclett.4c02555.
7. Mónica A. Gordillo, Mónica Soto‐Monsalve, Christian C. Carmona‐Vargas, Gustavo Gutiérrez, Richard F. D'vries, Jean‐Marie Lehn, Manuel N. Chaur. (2017). Photochemical and Electrochemical Triggered Bis(hydrazone) Switch. Chemistry – A European Journal, 23(59), p.14872. https://doi.org/10.1002/chem.201703065.
8. Harok Jeong, Sangmin Park, Junghoon Yang, Hye-Min Lee, Sangmin An, Yasuhiro Yamada, Jungpil Kim. (2023). Spectroscopic distinction of carbon nanobelts and nanohoops. Carbon, 201, p.829. https://doi.org/10.1016/j.carbon.2022.09.063.
Dimensions
PlumX
Visitas a la página del resumen del artículo
Descargas
Licencia
Los autores/as conservarán sus derechos de autor y garantizarán a la revista el derecho de primera publicación de su obra, el cuál estará simultáneamente sujeto a la Licencia de reconocimiento de Creative Commons (CC. Atribución 4.0) que permite a terceros compartir la obra siempre que se indique su autor y su primera publicación en esta revista.
Los autores/as podrán adoptar otros acuerdos de licencia no exclusiva de distribución de la versión de la obra publicada (p. ej.: depositarla en un archivo telemático institucional o publicarla en un volumen monográfico) siempre que se indique la publicación inicial en esta revista.
Se permite y recomienda a los autores/as difundir su obra a través de Internet (p. ej.: en archivos telemáticos institucionales o en su página web) antes y durante el proceso de envío, lo cual puede producir intercambios interesantes y aumentar las citas de la obra publicada. (Véase El efecto del acceso abierto).










