



Publicado
Validación de un método multirresiduo para la determinación de medicamentos veterinarios en trucha y langostino
Validation of a multiresidue method for the determination of veterinary drugs in rainbow trout and shrimp
Validação de um método de multiresíduos para la determinação o medicamentos veterinários em truta e camarão
DOI:
https://doi.org/10.15446/rev.colomb.quim.v51n2.101096Palabras clave:
multirresiduos, residuos de medicamentos veterinarios, UPLC-MS/MS, truchas, langostinos (es)Veterinary drug residues, UPLC/MS/MS, rainbow trouts, shrimps, multi-waste (en)
multi-resíduos, resíduos de medicamentos veterinários, UPLC-MS/MS, truta, camarão (pt)
Este estudio se realizó con el objetivo de desarrollar y validar un método para la determinación de 30 medicamentos veterinarios en muestras de trucha y langostino. El método utiliza extracción en fase sólida dispersiva (dSPE) con C18 y detección por cromatografía líquida acoplada a espectrometría de masas. Se determinó linealidad, veracidad (porcentaje de recuperación), repetitividad y reproducibilidad intralaboratorio (porcentaje de desviación estándar relativa (% RSD)), límites de detección (LoD), límites de cuantificación (LoQ), selectividad e incertidumbre. La recuperación varió de 70 a 120% y la repetibilidad y la reproducibilidad fueron menores de 20% de la desviación estándar relativa. La selectividad fue adecuada, sin picos interferentes. Las relaciones iónicas cumplieron con los criterios de confirmación. Los coeficientes de determinación (R2) fueron mayores de 0,99, con excepción de la sulfaquinoxalina en langostino (R2 = 0,97). Los LoD y los LoQ variaron entre 0,6 µg/kg y 12,8 µg/kg y los valores de incertidumbre entre 6 µg/kg y 49 µg/kg. Se analizaron adicionalmente 6 muestras de diferentes mercados de Lima y se detectaron trazas de algunos medicamentos incluidos en el ensayo. El método es adecuado para el análisis de residuos de medicamentos veterinarios y se recomienda su aplicación en los programas nacionales de monitoreo de la inocuidad de truchas y langostinos provenientes de acuicultura.
The study was aimed at developing and validate an analysis method to determine residues of 30 veterinary drugs in rainbow trout and shrimp specimens. The method involves extraction in dispersive solid phase with C18 and the subsequent detection through liquid chromatography coupled to mass spectrometry. Validation was done through determination of linearity, trueness (% of recovery), repeatability and intralaboratory reproducibility, limits of detection (LoD), limits of quantification (LoQ) selectivity and uncertainty. Recovery ranged from 70 to 120% and repeatability and intralaboratory reproducibility were lower than 20%. Selectivity was adequate, without interference peaks. Likewise, the ionic relationships met the confirmation criteria. The linearity was adequate, with determination coefficients (R2) above 0.99, except for sulfaquinolaxin in shrimp specimens (R2 = 0,97). LoD and LoQ varied from 0,6 µg/kg to 12,8 µg/kg. Limits of uncertainty ranged from 6 µg/kg to 49 µg/kg. The method was used to analyze 6 samples from different markets in Lima (Peru), identifying traces of some drugs included in the study. Our results show that the method is adequate for the analysis of veterinary drug residues and allow us to recommend its application in national monitoring programs, to assess the safety of rainbow trout and shrimp specimens from aquaculture.
The study was aimed at developing and validate an analysis method to determine residues of 30 veterinary drugs in rainbow trout and shrimp specimens. The method involves extraction in dispersive solid phase with C18 and the subsequent detection through liquid chromatography coupled to mass spectrometry. Validation was done through determination of linearity, trueness (% of recovery), repeatability and intralaboratory reproducibility, limits of detection (LoD), limits of quantification (LoQ) selectivity and uncertainty. Recovery ranged from 70 to 120% and repeatability and intralaboratory reproducibility were lower than 20%. Selectivity was adequate, without interference peaks. Likewise, the ionic relationships met the confirmation criteria. The linearity was adequate, with determination coefficients (R2) above 0.99, except for sulfaquinolaxin in shrimp specimens (R2 = 0,97). LoD and LoQ varied from 0,6 µg/kg to 12,8 µg/kg. Limits of uncertainty ranged from 6 µg/kg to 49 µg/kg. The method was used to analyze 6 samples from different markets in Lima (Peru), identifying traces of some drugs included in the study. Our results show that the method is adequate for the analysis of veterinary drug residues and allow us to recommend its application in national monitoring programs, to assess the safety of rainbow trout and shrimp specimens from aquaculture.
Aplicada y Analítica
Validación de un método multirresiduo para la determinación de medicamentos veterinarios en trucha y langostino
Validation of a multiresidue method for the determination of veterinary drugs in rainbow trout and shrimp
Validação de um método de multiresíduos para la determinação o medicamentos veterinários em truta e camarão
Validación de un método multirresiduo para la determinación de medicamentos veterinarios en trucha y langostino
Revista Colombiana de Química, vol. 51, núm. 2, pp. 16-24, 2022
Universidad Nacional de Colombia
Recepción: 16 Febrero 2022
Revisado: 08 Agosto 2022
Aprobación: 18 Agosto 2022
Resumen: Este estudio se realizó con el objetivo de desarrollar y validar un método para la determinación de 30 medicamentos veterinarios en muestras de trucha y langostino. El método utiliza extracción en fase sólida dispersiva (dSPE) con C18 y detección por cromatografía líquida acoplada a espectrometría de masas. Se determinó linealidad, veracidad (porcentaje de recuperación), repetitividad y reproducibilidad intralaboratorio (porcentaje de desviación estándar relativa (% RSD)), límites de detección (LoD), límites de cuantificación (LoQ), selectividad e incertidumbre. La recuperación varió de 70 a 120% y la repetibilidad y la reproducibilidad fueron menores de 20% de la desviación estándar relativa. La selectividad fue adecuada, sin picos interferentes. Las relaciones iónicas cumplieron con los criterios de confirmación. Los coeficientes de determinación (R2) fueron mayores de 0,99, con excepción de la sulfaquinoxalina en langostino (R2 = 0,97). Los LoD y los LoQ variaron entre 0,6 µg/kg y 12,8 µg/kg y los valores de incertidumbre entre 6 µg/kg y 49 µg/kg. Se analizaron adicionalmente 6 muestras de diferentes mercados de Lima y se detectaron trazas de algunos medicamentos incluidos en el ensayo. El método es adecuado para el análisis de residuos de medicamentos veterinarios y se recomienda su aplicación en los programas nacionales de monitoreo de la inocuidad de truchas y langostinos provenientes de acuicultura.
Palabras clave: multirresiduos, residuos de medicamentos veterinarios, UPLC-MS, MS, truchas, langostinos.
Abstract: This study was aimed at developing and validate an analysis method to determine residues of 30 veterinary drugs in rainbow trout and shrimp specimens. The method involves extraction in dispersive solid phase with C18 and the subsequent detection through liquid chromatography coupled to mass spectrometry. Validation was done through determination of linearity, trueness (% of recovery), repeatability and intralaboratory reproducibility, limits of detection (LoD), limits of quantification (LoQ) selectivity and uncertainty. Recovery ranged from 70 to 120% and repeatability and intralaboratory reproducibility were lower than 20%. Selectivity was adequate, without interference peaks. Likewise, the ionic relationships met the confirmation criteria. The linearity was adequate, with determination coefficients (R2) above 0.99, except for sulfaquinolaxin in shrimp specimens (R2 = 0,97). LoD and LoQ varied from 0,6 µg/kg to 12,8 µg/kg. Limits of uncertainty ranged from 6 µg/kg to 49 µg/kg. The method was used to analyze 6 samples from different markets in Lima (Peru), identifying traces of some drugs included in the study. Our results show that the method is adequate for the analysis of veterinary drug residues and allow us to recommend its application in national monitoring programs, to assess the safety of rainbow trout and shrimp specimens from aquaculture.
Keywords: Veterinary drug residues, UPLC, MS, MS, rainbow trouts, shrimps.
Resumo: O estudo foi realizado com o objetivo de desenvolver e validar um método para a determinação de 30 medicamentos veterinários, em amostras de truta e camarão. O método utiliza extração dispersiva em fase sólida com C18 e detecção por cromatografia líquida acoplada à espectrometria de massas. Foram determinados a linearidade, a veracidade (recuperação percentual), a repetibilidade, a reprodutibilidade intra-laboratorial, os limites de detecção (LoD) e de quantificação (LoQ), a linearidade, a selectividade e a incerteza. A recuperação variou de 70 a 120%, a repetibilidade e reprodutibilidade estiveram abaixo do 20% do desvio padrão relativo. A selectividade fio adequada, sem picos de interferentes. As proporções de íons atenderam aos critérios de confirmação. Os coeficientes de determinação (R2) foram superiores a 0,99, com excepção da sulfanoxalina em camarão (R2 = 0,97). LoD e LoQ variavam entre 0,6 µg/kg e 12,8 µg/kg e valores de incerteza entre 6 µg/kg e 49 µg/kg. Seis amostras de mercados do Lima foram adicionalmente analisadas e foram detectados vestígios de alguns medicamentos incluídos no estudo. O método é adequado para o análise de resíduos de medicamentos veterinários e sua aplicação é recomendada em programas nacionais de controlo da segurança da truta e do camarão provenientes da aquicultura.
Palavras-chave: multi-resíduos, resíduos de medicamentos veterinários, UPLC-MS, MS, truta, camarão.
Introducción
La acuicultura mundial muestra un incremento anual sostenido, según los reportes de la Organización de las Naciones Unidas para la Alimentación y la Agricultura [1]. La magnitud de extracción de productos provenientes de la pesca es oscilante, con épocas de mayor o menor biomasa marina extraída, en tanto, la producción proveniente de la acuicultura es más predecible, puesto que es controlada y el proceso de cría y su rendimiento se pueden modificar a voluntad [2]. En el Perú, las exportaciones acuícolas crecieron un 25,3% en el periodo enero-octubre de 2021, hasta alcanzar 338 millones de dólares estadounidenses y 43.691 toneladas [3].
Sin embargo, los productos acuícolas pueden contener residuos o contaminantes químicos, tales como residuos de medicamentos veterinarios, resultantes del uso intencionado (administración de fármacos para contrarrestar enfermedades) o no intencionado (contaminación ambiental), lo que genera un problema de salud pública y al mismo tiempo pone en riesgo la comercialización de los productos acuícolas, sobre todo en el mercado internacional [3], [4].
Medicamentos veterinarios tales como antibióticos y antiparasitarios cumplen un rol importante en la acuicultura para el tratamiento y prevención de enfermedades transmisibles. Sin embargo, cuando su uso es indiscriminado, cuando no se cumplen los periodos de retiro y las dosis establecidas, o cuando se utilizan sustancias no autorizadas, se pueden originar residuos de estas sustancias en los tejidos comestibles, que pueden causar efectos adversos en la salud humana, desde leves reacciones alérgicas hasta resistencia bacteriana y graves efectos cancerígenos o teratógenos [5], [6]. Por ello, las agencias gubernamentales de inocuidad alimentaria y en el ámbito internacional agencias como el Codex Alimentarius han establecido las concentraciones o límites máximos de residuos (LMR) que debe contener un alimento para ser considerado inocuo para el consumo humano [7]-[10]. Los exportadores de productos acuícolas deben cumplir con los requisitos sanitarios de LMR, establecidos por las autoridades de los países de destino, para evitar posibles detenciones y rechazos de sus envíos debido a la presencia de residuos o contaminantes en cantidades superiores a las permitidas. Los LMR de los medicamentos veterinarios incluidos en el presente estudio varían desde 100 hasta 2000 µg/kg; asimismo, el cloranfenicol y los beta-agonistas se encuentran prohibidos para uso en la producción animal, según lo establecido por el Código de Regulación de los Estados Unidos [7], por la Regulación Europea [8], [9] y por el Codex Alimentarius [10].
Los laboratorios que realizan ensayos de residuos de medicamentos veterinarios juegan un rol importante en el control de la inocuidad alimentaria de productos de acuicultura en el mercado nacional e internacional. En el Perú, la entidad encargada de realizar los controles oficiales de residuos de medicamentos veterinarios en los productos acuícolas es el Organismo Nacional de Sanidad Pesquera, que realiza muestreos en centros de producción y en lotes de exportación, y envía las muestras a laboratorios extranjeros para su análisis, debido a la no disponibilidad de laboratorios nacionales que realicen estos ensayos.
La cromatografía líquida acoplada a espectrometría de masas en tándem (UPLC-MS/MS) es la técnica de referencia recomendada por la Unión Europea [11], [12] y por la guía de validación de la administración de alimentos y medicamentos de los Estados Unidos [13] para realizar los análisis de residuos de medicamentos veterinarios en los alimentos, debido a su alta sensibilidad y selectividad, que permiten realizar el estudio simultáneo de diferentes grupos de fármacos y/o sus metabolitos en niveles de trazas, en el orden de partes por billón y en concentraciones menores de los LMR. La UPLC-MS/MS genera información de la estructura molecular de los analitos, a partir del monitoreo de los iones productos originados por la disociación de la molécula precursora, que se utiliza para confirmar la identidad de los analitos [11]–[13]. Esta característica de la UPLC-MS/MS ayuda a evitar o disminuir la ocurrencia de resultados falsos positivos o falsos negativos, que pueden originar problemas de detenciones o rechazos de los envíos, así como pérdidas económicas y disputas legales en el comercio internacional de alimentos. Además, la UPLC-MS/MS permite realizar análisis multirresiduo para la detección y cuantificación simultánea de cientos de moléculas en una misma corrida, sin necesidad de una rigurosa separación cromatográfica, lo cual simplifica los procesos analíticos y facilita una rápida respuesta de los laboratorios de ensayo en el control de la inocuidad alimentaria [11].
La interferencia de matriz es una de las principales desventajas de la técnica UPLC-MS/MS, que se origina por la presencia de ciertos compuestos orgánicos de los alimentos que permanecen como impurezas en los extractos de las muestras y afectan la ionización de los analitos. Para superar esta interferencia se requiere realizar una exhaustiva limpieza de los extractos, mediante uno o más métodos de purificación, tales como la extracción en fase sólida, extracción líquido-líquido, extracción asistida por microonda o extracción acelerada por solventes. Sin embargo, los laboratorios que realizan análisis rutinarios requieren utilizar métodos sencillos con la finalidad de generar resultados rápidos para el control de la inocuidad alimentaria. La dSPE, introducida para el análisis de residuos de plaguicidas en frutos y vegetales [14], es actualmente la técnica de extracción y limpieza de muestras de mayor aceptación internacional en los laboratorios que realizan análisis para el control de la inocuidad alimentaria.
El presente estudio se realizó con el objetivo de desarrollar y validar un método de análisis sencillo, robusto y efectivo para la determinación multirresiduo de 30 medicamentos veterinarios pertenecientes a diferentes familias de fármacos (penicilinas, tetraciclinas, macrolidos, entre otros), en trucha (Oncorhynchus mykiss) y langostino (Litopenaeus vannamei). El método presentado en este artículo utiliza dSPE con C18 y un posterior análisis instrumental por UPLC-MS/MS.
Materiales y métodos
Estándares y reactivos
Para evaluar los medicamentos veterinarios de interés se utilizaron estándares de alta pureza (mayor de 99%). Los estándares de tetraciclina, clortetraciclina, oxitetraciclina, ácido oxolínico, flumequina, ácido nalidíxico, enrofloxacino, ciprofloxacino, sulfamerazina, sulfacloropiridazina, sulfadiazina, sulfametoxazol, sulfaquinolaxina, sulfametazina, sulfatiazol, sulfadimetoxina, sulfametoxipiridazina, sulfadoxina, espiramicina, eritromicina, trimetoprim, cimaterol, salbutamol, Terbutalina, ractopamina, zilpaterol, clembuterol, florfenicon y cloranfenicol fueron adquiridos de Sigma Aldrich (San Luis, EE. UU.). Los estándares de amoxicilina y mapenterol fueron adquiridos de Dr. Ehrenstorfer (Augsburg, Alemania).
Los reactivos utilizados fueron de grado LC-MS o HPLC. Metanol y acetonitrilo fueron obtenidos de Sigma-Aldrich, ácido fórmico de Fluka, sulfato de magnesio anhidro y acetato de sodio anhidro de JT Baker (Deventer, Holland) y agua ultrapura del equipo Milipore Mili-Q system (Milford, MA, EE. UU.).
Las soluciones individuales concentradas de 2000 ng/µL (soluciones stock) de cada estándar de medicamento fueron preparadas disolviendo una cantidad apropiada de cada estándar primario en acetonitrilo, metanol o agua ultrapura. Las soluciones fueron almacenadas a -20 °C en la oscuridad. Las soluciones de mezclas estándares de 40 ng/µL fueron preparadas tomando una alícuota de cada una de las soluciones stock y disolviéndolas en acetonitrilo, con excepción de los betalactámicos, en los cuales se utilizó agua para la disolución y almacenamiento en viales plásticos, a fin de evitar problemas de absorción en vidrio de los antibióticos. Las soluciones de calibración de 10 ng/µL y 2 ng/µL fueron preparadas disolviendo en acetonitrilo una cantidad apropiada de las soluciones de mezclas de estándares de 40 ng/µL.
Preparación de las muestras
Las muestras de truchas y langostinos fueron obtenidas de empresas locales dedicadas a la acuicultura. Fueron mantenidas en almacenamiento a -20 °C y protegidas de la luz hasta su preparación para el análisis. Así mismo, fueron descongeladas y llevadas a temperatura ambiente antes de realizar los ensayos. Se procedió a la molienda de langostinos (sin cabeza y antenas) y trucha (solo músculo) utilizando el homogenizador Robot coupe Blixer 3.0. Luego se realizó la extracción de la siguiente manera: se pesaron aproximadamente 2,00 g de músculo de trucha o langostino dentro de tubos de centrifuga de polipropileno de 50 mL, luego se agregaron 10 mL de acetonitrilo/EDTA 0.1 M en agua (4/1%v/v) y se homogeneizó con un equipo vortex por 5 min, luego se procedió a centrifugar por 5 min a 4000 rpm, el sobrenadante se trasvasó a un tubo de 50 mL que contenía 300 mg de adsorbente C18; luego se adicionaron 10 mL de hexano presaturado en acetonitrilo, se agitó por 30 s y se procedió a centrifugar nuevamente por 5 min a 4000 rpm. Se eliminó la capa de hexano con una pipeta Pasteur, del sobrenadante que quedó se tomó una alícuota de 5 mL y se llevó a un tubo de centrifuga de polipropileno de 15 mL. Finalmente, se concentró el extracto a sequedad, en un equipo de evaporación de nitrógeno a 45 °C. El extracto evaporado fue reconstituido con 1 mL de fase móvil A (ácido fórmico al 0,1% en agua), se filtró a través de filtros de difluoruro de polivinilo (PDVF) de 0,2 µm y se colocó en viales de color ámbar. Finalmente, se inyectaron 2 µL en el equipo de UPLC-MS/MS.
Análisis por UPLC-MS/MS
El análisis por cromatografía se realizó con el equipo Acquity UPLC system (Waters, Milford, MA, EE. UU.) y la separación de los analitos se realizó con la columna Acquity UPLC BEH C18 (100 mm x 2,1 mm, 1,7 µm). Para la corrida cromatográfica se utilizó como fase móvil ácido fórmico al 0,1% en agua ultrapura (Fase A) y 0,1% ácido fórmico en acetonitrilo (Fase B) con un flujo de 0,5 mL/min. El gradiente de elución inició con 0% fase móvil B por 0,1 min, que se incrementó en forma lineal hasta 100% en 7,9 min, y se mantuvo por 1,5 min antes de regresar a la condición inicial en 0,1 min, con un tiempo de preequilibración de 3,4 min. El tiempo de la corrida cromatográfica fue de 13 min, el volumen de inyección fue de 2 µL y la temperatura de la columna fue de 40 °C.
El análisis por espectrometría de masas se realizó con el equipo UPLC-MS/MS Acquity Xevo TQ-XS (Waters, Manchester, Reino Unido). El equipo fue operado utilizando la fuente de ionización por electrospray (ESI) en modo ESI positivo. Los parámetros para la fuente de ionización fueron: voltaje de capilar 3,0 kV, temperatura de la fuente 150 °C, temperatura de desolvatación 500 °C, flujo del gas de cono 150 L/h y flujo del gas de desolvatación 1000 L/h. La colisión inducida por disociación se realizó utilizando argón como gas de colisión, a una presión 4 x10-3
mbar en la celda de colisión. La adquisición y procesamiento de datos en el equipo UPLC-MS/MS se realizó con el programa MassLynx 4.2. En la Tabla 1 se muestran los principales parámetros de espectrometría de masa MS/MS por cada analito determinado.
| Analito | Grupo farmacológico | Ventana tR (min) | Cono (V) | Ion precursor (m/z) | Iones productos (m/z) | ||||
| Cuantia (m/z) | ECb (eV) | Cualic (m/z) | EC (eV) | ||||||
| 1 | Oxitetraciclina | Tetraciclinas | 1,98 – 2,78 | 40 | 461v3 | 426,3 | 18 | 443,3 | 12 |
| 2 | Clortetraciclina | Tetraciclinas | 2,59 – 3,39 | 20 | 479,4 | 444,2 | 20 | 462,2 | 15 |
| 3 | Tetraciclina | Tetraciclinas | 2,11 – 2,91 | 20 | 445,3 | 410,3 | 18 | 153,7 | 25 |
| 4 | Enrofloxacino | Quinolonas | 2,14 – 2,94 | 52 | 360,2 | 316,2 | 18 | 245 | 24 |
| 5 | Flumequina | Quinolonas | 3,79 – 4,59 | 40 | 262 | 244 | 18 | 201,8 | 32 |
| 6 | Ciprofloxacino | Quinolonas | 1,98 – 2,78 | 50 | 332,3 | 288,1 | 16 | 244,9 | 22 |
| 7 | Ácido nalidíxico | Quinolonas | 3,66 – 4,46 | 6 | 233 | 214,9 | 14 | 186,8 | 22 |
| 8 | Ácido oxolínico | Quinolonas | 3,05 – 3,85 | 24 | 262 | 244 | 18 | 159,7 | 34 |
| 9 | Sulfamerazina | Sulfonamidas | 2,04 – 2,84 | 34 | 265 | 91,6 | 26 | 155,7 | 16 |
| 10 | Sulfacloropiridazina | Sulfonamidas | 2,62 – 3,42 | 40 | 285,3 | 155,7 | 12 | 91,5 | 28 |
| 11 | Sulfadiazina | Sulfonamidas | 1,75 – 2,55 | 42 | 251 | 155,7 | 12 | 91,6 | 22 |
| 12 | Sulfametoxazol | Sulfonamidas | 2,78 – 3,58 | 10 | 254 | 155,7 | 14 | 91,5 | 24 |
| 13 | Sulfaquinolaxina | Sulfonamidas | 3,23 – 4,03 | 54 | 301,1 | 155,7 | 14 | 91,5 | 34 |
| 14 | Sulfametazina | Sulfonamidas | 2,29 – 3,09 | 42 | 279,1 | 185,8 | 16 | 123,7 | 25 |
| 15 | Sulfatiazol | Sulfonamidas | 1,84 – 2,64 | 38 | 256 | 155,7 | 15 | 91,5 | 26 |
| 16 | Sulfadimetoxina | Sulfonamidas | 3,22 – 4,02 | 56 | 311,1 | 155,7 | 22 | 91,5 | 32 |
| 17 | Sulfametoxipiridazina | Sulfonamidas | 2,30 – 3,10 | 46 | 281 | 155,7 | 15 | 91,5 | 26 |
| 18 | Sulfadoxina | Sulfonamidas | 2,77 – 3,57 | 44 | 311,1 | 155,7 | 16 | 91,5 | 26 |
| 19 | Cimaterol | B-agonistas | 1,33 – 2,13 | 22 | 220 | 159,8 | 16 | 142,7 | 23 |
| 20 | Salbutamol | B-agonistas | 1,27 – 2,07 | 34 | 240,1 | 147,7 | 18 | 165,8 | 12 |
| 21 | Terbutalina | B-agonistas | 1,26 – 2,06 | 16 | 226 | 151,8 | 14 | 106,6 | 28 |
| 22 | Ractopamina | B-agonistas | 2,04 – 2,84 | 35 | 302,2 | 163,8 | 15 | 284,2 | 12 |
| 23 | Zilpaterol | B-agonistas | 1,26 – 2,06 | 20 | 262,1 | 244,1 | 12 | 184,9 | 23 |
| 24 | Clembuterol | B-agonistas | 2,33 – 3,13 | 8 | 277 | 202,8 | 14 | 131,7 | 24 |
| 25 | Mapenterol | B-agonistas | 2,95 – 3,75 | 22 | 325,2 | 236,9 | 14 | 307,1 | 10 |
| 26 | Espiramicina | Macrólidos | 2,45 – 3,25 | 20 | 422,5 | 173,9 | 20 | 100,6 | 15 |
| 27 | Amoxicilina | Penicilinas | 1,26 – 2,06 | 6 | 366,3 | 349,2 | 6 | 113,5 | 18 |
| 28 | Florfenicol | Fenicoles | 2,82 – 3,62 | 25 | 358,2 | 240,8 | 16 | 205,8 | 26 |
| 29 | Trimetroprim | Trimetroprim | 1,88 – 2,68 | 40 | 291,1 | 230 | 22 | 122,6 | 28 |
| 30 | Cloranfenicol | Fenicoles | 3,01 – 3,81 | 30 | 323,2 | 275 | 12 | 164,8 | 22 |
bEnergía de colisión.
cIon producto segundo en abundancia, utilizado para la detección cualitativa.
Validación del método
La validación del método se realizó con la finalidad de verificar el funcionamiento analítico del método desarrollado para el uso propuesto, es decir, el análisis de residuos de medicamentos veterinarios en productos acuícolas (truchas y langostinos). La validación se realizó de acuerdo al procedimiento de la Regulación Unión Europea 2002/657 [11] y Eurachem [15]. Para ello, se utilizó muestra de blancos de matriz de truchas y langostinos fortificados con cantidades apropiadas de la solución de estándares de trabajo para obtener tres niveles de concentraciones de 10, 50 y 100 µg/kg, con 10 análisis repetidos por cada nivel. Para determinar la reproducibilidad intralaboratorio, se realizaron los análisis en tres ocasiones diferentes, con tres analistas. La linealidad fue evaluada utilizando curva de calibración fortificada en blanco de matriz en 5 niveles de concentración de 10 µg/kg hasta 300 µg/kg. Los límites de deteción (LoD) y de cuantificación (LoQ) fueron estimados utilizando blancos de matriz fortificados a la concentración de 10 µg/kg, con 10 análisis repetidos.
La evaluación de la selectividad del método por UPLC-MS/MS se realizó mediante adquisición por espectrometría de masa en modo de monitoreo de reacción múltiple (MRM), con dos iones productos por cada compuesto. La proporción de las señales de los iones productos (relaciones iónicas) de cada compuesto se halló dividiendo las señales del ion menos abundante entre el ion más abundante. La identificación de los compuestos por UPLC-MS/MS se realizó según los requisitos de relación iónica establecidos por Codex [13].
La estimación de la incertidumbre se realizó de acuerdo al procedimiento establecido en la Guía ISO o método GUM [16] y Eurachem [17]. La estimación de la incertidumbre combinada se calculó a partir de la Ec. (1):

Donde C=Ccontenido en la muestra en µg/kg, Co=concentración en el extracto en ng/mL obtenida por interpolación en la curva de calibración, Ve=volumen del extracto, W=peso de la muestra, F=factor de concentración y R=repetibilidad. La incertidumbre expandida se calculó multiplicando la incertidumbre combinada por K=2, considerando un nivel de confianza de 95%.
La evaluación de los parámetros de validación se realizó de acuerdo a los criterios de aceptación establecidos por el Codex Alimentarius [13], con RSD menor de 20% para la repetibilidad y reproducibilidad intralaboratorio y con recuperación de 70% a 120% para la veracidad. La selectividad del método se evaluó comparando los cromatogramas de los blancos de matriz versus los blancos de matriz fortificados, verificando que no hubiera señales interferentes en la región en donde eluyen los analitos. Asimismo, se realizó la confirmación por espectrometría de masas comparando la relación iónica de dos fragmentos MS/MS de cada molécula, verificando que las relaciones iónicas de las muestras estuvieran dentro del intervalo de aceptabilidad, en comparación con los estándares establecidos por el Codex Alimentarius [13].
Resultados y discusión
Análisis por UPLC-MS/MS
La optimización de los parámetros de espectrometría de masa MS/MS fue realizada mediante infusión directa en el espectrómetro de soluciones de cada uno de los estándares de medicamentos veterinarios, a la concentración de 0,5 mg/L. El electrospray fue utilizado en modo ion positivo, puesto que todos los analitos presentaban mayor señal en este modo de ionización. En la Tabla 1 se muestran las condiciones del espectrómetro de masas utilizadas en el estudio. En cuanto a la condiciones cromatográficas, fueron desarrolladas principalmente para optimizar la forma de pico, resolución e intensidad de los analitos. La fase móvil fue investigada principalmente para maximizar la sensibilidad y la resolución del método. Se probaron diferentes solventes polares, tales como metanol o acetonitrilo y agua ultrapura, todos con concentraciones de ácido fórmico al 0,05 y 0,1% (v/v). El acetonitrilo presentó mayor sensibilidad para los analitos y el ácido fórmico favoreció la ionización de los analitos. De igual manera, se configuró el tiempo de monitoreo de cada transición (dwell time) utilizando el programa MassLynx 4.2, con la finalidad de obtener como mínimo 15 puntos en cada pico cromatográfico.
En la Figura 1 se muestra el cromatograma de ion total (TIC) para la matriz trucha. Los tiempos de retención de los compuestos en las matrices de trucha y langostino fueron similares; variaron desde 1,66 hasta 4,19 minutos. La gradiente utilizada sirvió para la separación de los analitos en menos de 5 minutos. A pesar de que algunos compuestos coeluyen, ello no constituyó problema para su cuantificación, debido a la alta selectividad de la técnica de UPLC-MS/MS, puesto que estos compuestos son separados y detectados mediante adquisición de monitoreo de reacción múltiple por espectrometría de masas en tándem.
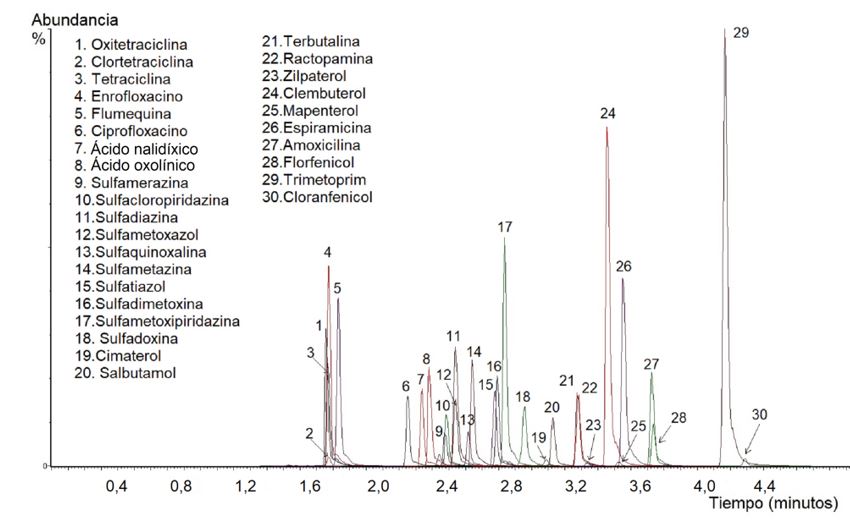
Figura 1. Cromatogramas de ion total (TIC) de 30 residuos de medicamentos veterinarios, fortificados a 100 µg/kg en blanco de matriz de trucha.
Validación del método
Después de determinar las condiciones de extracción, de separación cromatográfica y de detección por espectrometría de masas, se validó el método de análisis, determinándose los parámetros de recuperación, repetitividad, reproducibilidad intralaboratorio, linealidad, LoD, LoQ, selectividad e incertidumbre.
Recuperación
En las Tablas 2 y 3 se observa que los porcentajes de recuperación variaron desde 76,8 hasta 120,5%, valores que se encuentran dentro de la especificación establecida por el Codex Alimentarius, es decir, entre 70 y 120%.
| Analito | 10 µg/kg | 50 µg/kg | 100 µg/kg | |||||||
| Ra | RSDrb | RSDRc | R | RSDr | RSDR | R | RSDr | RSDR | ||
| 1 | Oxitetraciclina | 100 | 1,9 | 5,4 | 91 | 11,0 | 11,5 | 93 | 2,3 | 14,1 |
| 2 | Clortetraciclina | 104 | 7,1 | 7,0 | 96 | 4,9 | 4,2 | 98 | 4,0 | 5,5 |
| 3 | Tetraciclina | 98 | 4,0 | 6,0 | 91 | 12,8 | 12,6 | 93 | 1,8 | 13,8 |
| 4 | Enrofloxacino | 93 | 2,8 | 8,7 | 97 | 8,8 | 10,4 | 99 | 2,1 | 12,1 |
| 5 | Flumequina | 106 | 3,6 | 4,1 | 93 | 9,5 | 10,6 | 98 | 1,0 | 10,1 |
| 6 | Ciprofloxacino | 93 | 5,7 | 5,3 | 96 | 9,1 | 9,5 | 97 | 2,2 | 10,1 |
| 7 | Ácido nalidíxico | 95 | 1,5 | 2,2 | 95 | 8,6 | 10,9 | 99 | 1,1 | 11,6 |
| 8 | Ácido oxolínico | 91 | 3,5 | 3,4 | 98 | 9,9 | 11,2 | 101 | 1,7 | 11,2 |
| 9 | Sulfamerazina | 89 | 3,9 | 5,3 | 101 | 7,2 | 9,2 | 107 | 3,3 | 9,3 |
| 10 | Sulfacloropiridazina | 99 | 3,1 | 5,9 | 95 | 5,7 | 7,4 | 100 | 2,4 | 9,5 |
| 11 | Sulfadiazina | 100 | 2.8 | 4,8 | 95 | 12,7 | 12,8 | 99 | 1,8 | 12,5 |
| 12 | Sulfametoxazol | 100 | 2,3 | 3,4 | 96 | 4,0 | 6,4 | 101 | 2,0 | 7,0 |
| 13 | Sulfaquinoxalina | 98 | 3,3 | 5,6 | 92 | 9,1 | 10,9 | 98 | 1,6 | 10,3 |
| 14 | Sulfametazina | 100 | 2,9 | 3,2 | 90 | 11,2 | 11,2 | 96 | 1,6 | 11,6 |
| 15 | Sulfatiazol | 96 | 2,8 | 5,2 | 92 | 9,3 | 10,4 | 98 | 1,6 | 11,0 |
| 16 | Sulfadimetoxina | 96 | 3,6 | 6,8 | 89 | 10,7 | 14,0 | 95 | 1,3 | 15,0 |
| 17 | Sulfametoxipiridazina | 103 | 1,6 | 3,4 | 95 | 10,4 | 12,0 | 101 | 2,8 | 12,0 |
| 18 | Sulfadoxina | 103 | 3,0 | 3,5 | 95 | 5,2 | 7,2 | 99 | 1,9 | 8,5 |
| 19 | Cimaterol | 77 | 2,8 | 3,6 | 98 | 5,6 | 7,1 | 101 | 1,6 | 8,9 |
| 20 | Salbutamol | 84 | 1,5 | 3,3 | 108 | 2,7 | 3,0 | 106 | 1,0 | 4,4 |
| 21 | Terbutalina | 82 | 4,2 | 4,5 | 106 | 2,2 | 3,0 | 104 | 1,8 | 4,1 |
| 22 | Ractopamina | 88 | 2,8 | 9,9 | 109 | 2,3 | 3,7 | 109 | 1,1 | 4,1 |
| 23 | Zilpaterol | 93 | 3,1 | 3,5 | 101 | 4,6 | 4,4 | 100 | 1,6 | 6,1 |
| 24 | Clembuterol | 96 | 1,9 | 2,8 | 103 | 2,3 | 1,9 | 106 | 1,5 | 2,7 |
| 25 | Mapenterol | 92 | 3,2 | 4,1 | 99 | 2,3 | 2,8 | 103 | 1,4 | 5,0 |
| 26 | Espiramicina | 100 | 3,3 | 3,3 | 98 | 2,8 | 3,0 | 103 | 2,7 | 4,8 |
| 27 | Amoxicilina | 103 | 3,5 | 4,4 | 95 | 4,3 | 3,7 | 96 | 2,1 | 6,4 |
| 28 | Florfenicol | 99 | 5,8 | 7,2 | 97 | 6,2 | 6,3 | 100 | 1,9 | 8,0 |
| 29 | Trimetoprim | 89 | 2,1 | 4,9 | 111 | 2,5 | 2,5 | 110 | 1,4 | 3,9 |
| 30 | Cloranfenicol | 90 | 6,4 | 7,1 | 94 | 8,4 | 8,1 | 98 | 3,4 | 11,2 |
| aRecuperación de 10 análisis en 03 ocasiones (n=30) (%). | ||||||||||
| bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%). | ||||||||||
| cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%). | ||||||||||
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).
| Analito | 10 µg/kg | 50 µg/kg | 100 µg/kg | |||||||
| Ra | RSDrb | RSDRc | R | RSDr | RSDR | R | RSDr | RSDR | ||
| 1 | Oxitetraciclina | 113 | 2,4 | 4,4 | 80 | 2,2 | 6,3 | 100 | 1,6 | 8,1 |
| 2 | Clortetraciclina | 116 | 4,6 | 4,3 | 81 | 9,0 | 9,8 | 97 | 6,1 | 6,9 |
| 3 | Tetraciclina | 115 | 2,6 | 3,2 | 82 | 3,8 | 4,6 | 99 | 1,8 | 5,4 |
| 4 | Enrofloxacino | 113 | 2,1 | 3,4 | 82 | 2,7 | 5,1 | 97 | 2,0 | 6,2 |
| 5 | Flumequina | 109 | 4,7 | 6,5 | 79 | 4,7 | 3,7 | 97 | 1,6 | 3,3 |
| 6 | Ciprofloxacino | 109 | 5,1 | 7,1 | 80 | 4,1 | 5,9 | 96 | 1,9 | 6,5 |
| 7 | Ácido nalidíxico | 112 | 2,5 | 3,4 | 82 | 2,1 | 2,6 | 100 | 1,0 | 2,8 |
| 8 | Ácido oxolínico | 113 | 1,9 | 3,0 | 84 | 1,5 | 2,5 | 103 | 1,8 | 3,6 |
| 9 | Sulfamerazina | 92 | 2,8 | 5,2 | 99 | 2,1 | 3,5 | 111 | 1,9 | 4,5 |
| 10 | Sulfacloropiridazina | 114 | 2,8 | 4,5 | 82 | 2,2 | 4,2 | 98 | 1,4 | 5,2 |
| 11 | Sulfadiazina | 110 | 3,1 | 4,8 | 88 | 2,2 | 2,1 | 101 | 2,5 | 2,4 |
| 12 | Sulfametoxazol | 117 | 3,4 | 2,4 | 83 | 2,6 | 2,6 | 99 | 1,4 | 1,9 |
| 13 | Sulfaquinoxalina | 116 | 3,2 | 6,1 | 79 | 1,9 | 3,6 | 101 | 1,5 | 4,2 |
| 14 | Sulfametazina | 102 | 3,4 | 4,6 | 91 | 3,4 | 3,4 | 107 | 1,2 | 3,2 |
| 15 | Sulfatiazol | 107 | 2,2 | 2,7 | 85 | 2,0 | 4,1 | 100 | 2,0 | 2,3 |
| 16 | Sulfadimetoxina | 113 | 0,8 | 2,9 | 81 | 1,6 | 3,9 | 99 | 1,6 | 3,9 |
| 17 | Sulfametoxipiridazina | 104 | 3,8 | 3,5 | 89 | 2,4 | 3,4 | 105 | 3,2 | 3,6 |
| 18 | Sulfadoxina | 115 | 2,4 | 2,4 | 84 | 1,1 | 2,2 | 100 | 2,1 | 3,9 |
| 19 | Cimaterol | 102 | 2,5 | 3,9 | 89 | 1,1 | 3,2 | 101 | 1,7 | 3,6 |
| 20 | Salbutamol | 111 | 1,8 | 3,4 | 85 | 0,6 | 1,9 | 96 | 1,0 | 2,2 |
| 21 | Terbutalina | 109 | 1,7 | 2,2 | 88 | 1,5 | 3,9 | 97 | 2,4 | 4,1 |
| 22 | Ractopamina | 115 | 2,1 | 4,4 | 81 | 2,1 | 3,6 | 101 | 1,4 | 5,0 |
| 23 | Zilpaterol | 114 | 1,4 | 4,4 | 86 | 1,9 | 4,4 | 99 | 1,2 | 4,1 |
| 24 | Clembuterol | 114 | 1,5 | 2,2 | 83 | 2,3 | 2,7 | 101 | 1,2 | 3,4 |
| 25 | Mapenterol | 112 | 1,4 | 3,0 | 82 | 1,7 | 3,6 | 99 | 2,3 | 4,8 |
| 26 | Espiramicina | 112 | 1,5 | 2,8 | 89 | 2,2 | 3,5 | 90 | 1,7 | 4,5 |
| 27 | Amoxicilina | 118 | 2,1 | 5,1 | 86 | 2,5 | 6,6 | 86 | 4,0 | 5,8 |
| 28 | Florfenicol | 121 | 9,0 | 10,6 | 88 | 6,3 | 6,0 | 103 | 3,6 | 4,8 |
| 29 | Trimetoprim | 107 | 2,0 | 4,0 | 87 | 2,3 | 3,5 | 104 | 2,1 | 4,1 |
| 30 | Cloranfenicol | 114 | 5,0 | 11,1 | 83 | 4,9 | 6,9 | 99 | 3,6 | 4,9 |
| aRecuperación de 10 análisis en 03 ocasiones (n=30) (%). | ||||||||||
| bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%). | ||||||||||
| cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%). | ||||||||||
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).
Repetibilidad y reproducibilidad intralaboratorio
En las Tablas 2 y 3 se observa que la repetibilidad y la reproducibilidad intralaboratorio, expresadas como % RSD, variaron desde 0,6% hasta 12,8%, con lo que se cumple con los criterios del Codex Alimentarius que establecen un % RSD < 20%. Cabe destacar que dichos porcentajes se encuentran en línea con otros estudios, en los cuales se validó la técnica analítica en matrices como pescados y langostinos, en los que se obtuvieron % RSD superiores a 20 % [18]-[21].
Límites de detección y de cuantificación
El LoD y el LoQ hallados en trucha y langostinos variaron desde 0,6 µg/kg hasta 12,8 µg/kg. En la Figura 2 se observa que la mayoría de los LoQ de los medicamentos incluidos fueron menores de 10 µg/kg, a excepción de cloranfenicol y florfenicol en langostino, que fueron de 12,7 y 12,8 µg/kg, respectivamente. Dichos valores están muy por debajo de los LMR de estos compuestos, que varían desde 50 hasta 2000 µg/kg, a excepción del cloranfenicol y los compuestos beta-agonistas, medicamentos prohibidos para uso en animales para consumo humano y para los que no se han establecido sus respectivos LMR.
Los LoQ hallados para cloranfenicol fueron de 6,4 y 12,7 µg/kg para trucha y langostino, respectivamente, superiores al límite de performance mínimo requerido de 0,3 µµg/kg establecido para cloranfenicol por la Regulación Europea [6], lo que se explicaría por el hecho de que, al haberse utilizado un método multirresiduo, no se puede aplicar condiciones de sintonización del espectrómetro de masas, condiciones cromatográficas y condiciones de extracción especificas con la finalidad de incrementar la sensibilidad del método para esta molécula. Por ello, lo recomendable sería realizar un método individual para el análisis del cloranfenicol, con la finalidad de obtener límites analíticos menores o iguales a lo requerido. Una situación diferente se observó para el florfenicol, para el cual se obtuvo LoQ de 7,2 y 12,8 µµg/kg para trucha y langostino, respectivamente, similares al cloranfenicol, por lo que estos límites analíticos obtenidos serian adecuados para el análisis de florfenicol, debido a que están muy por debajo del LMR, que es de 1000 µg/kg. Hay que remarcar que en investigaciones previas se han reportado valores de LoD y LoQ similares o por debajo de lo hallado en el presente estudio [18]-[26].
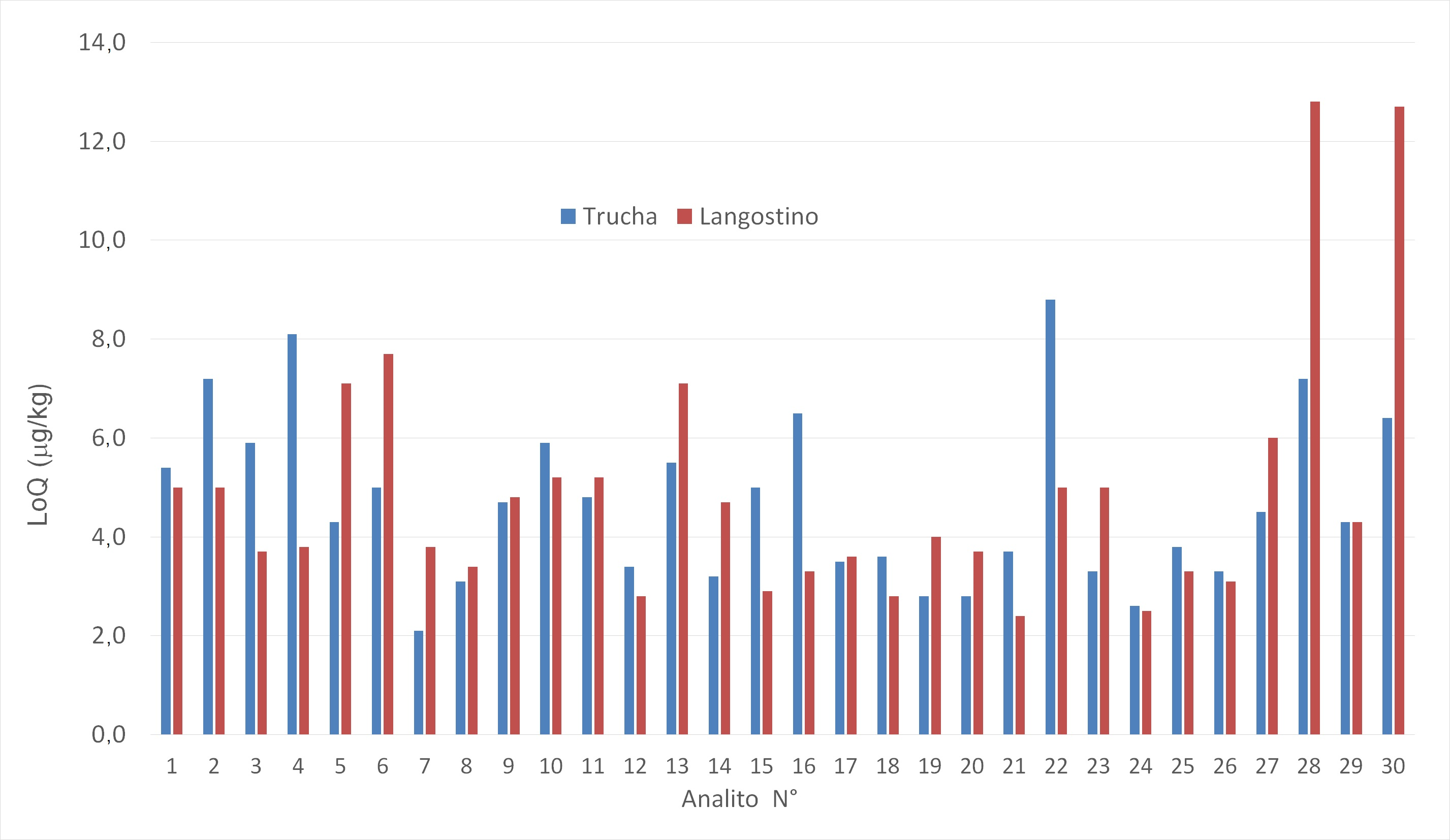
Figura 2.
Figura 2. Límite de cuantificación (LoQ) (µg/kg) del método en trucha y langostino.
Linealidad
Los coeficientes de determinación (R.) de las curvas de calibración de los medicamentos veterinarios fortificados en trucha y langostino fueron menores de 0,99; con excepción de sulfaquinoxalina en langostino, que fue de 0,97. En la Figura 3 se observan las curvas de calibración para la matriz trucha de compuestos representativos de los principales grupos de fármacos veterinarios incluidos en el estudio de validación.
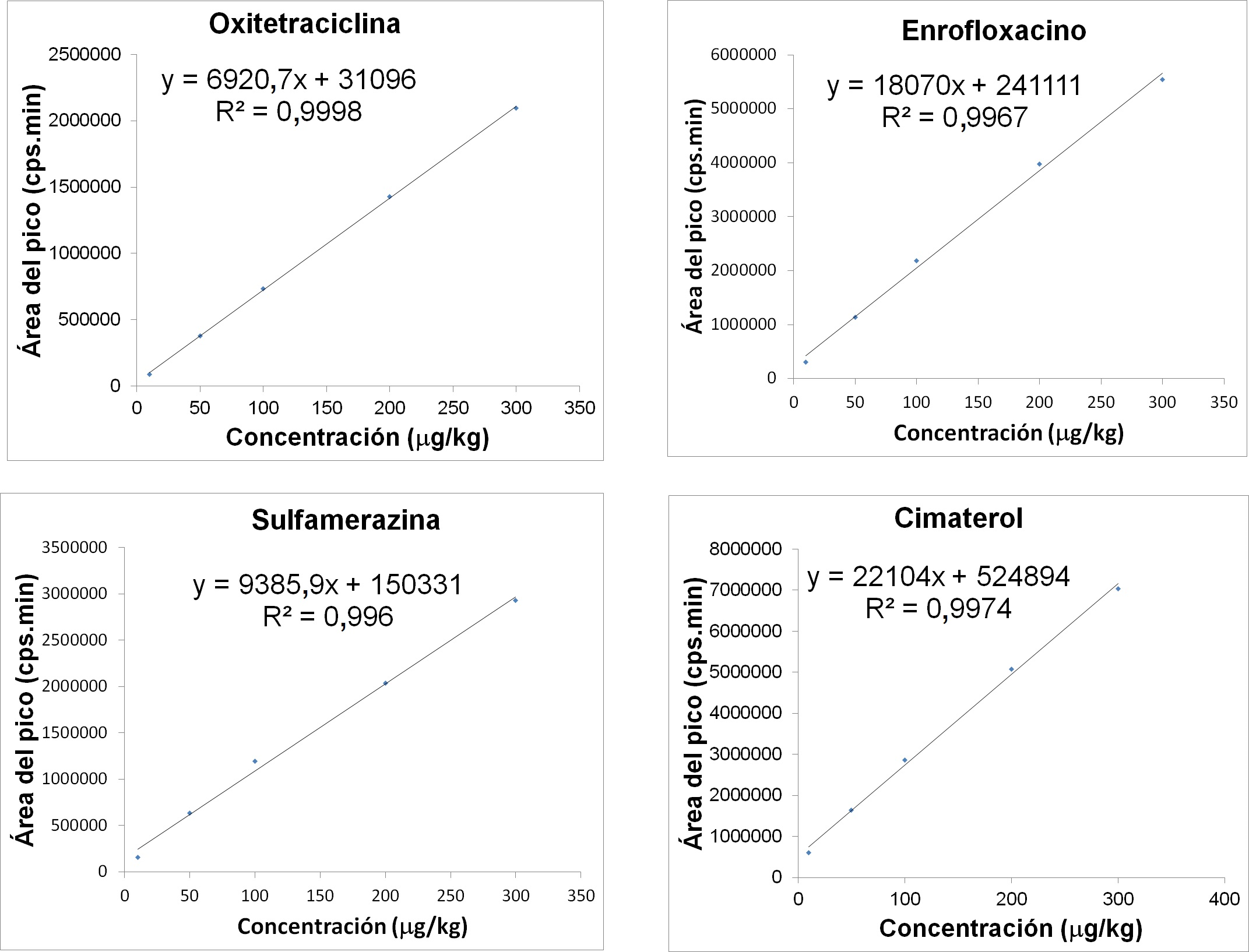
Figura 3
Figura 3. Curva de calibración de compuestos representativos fortificados en blanco de matriz de trucha.
Selectividad
La selectividad del método se evidenció mediante identificación por LC-MSMS de los medicamentos veterinarios incluidos en el estudio, dado que las relaciones iónicas de los compuestos cumplieron con los rangos de aceptación del Codex [13]. En la Figura 4 se muestran los cromatogramas MRM de los compuestos representativos fortificados en blando de matriz trucha, en los que se observa que el monitoreo de dos iones fragmentos característicos de la estructura molecular de cada compuesto le confiere una alta selectividad a la técnica de UPLC-MS/MS. Asimismo, se identificaron las moléculas por el tiempo de retención (TR). Por otro lado, se observó efecto de arrastre en los compuestos del grupo de las quinolonas, vale decir, ácido nalidíxico, ácido oxolínico, enrofloxacina y ciprofloxacina. Para superar este efecto, se requeriría investigar los métodos de lavado del inyector utilizando diferentes solventes y tiempos de lavado. Sin embargo, debe puntualizarse que el efecto de arrastre fue insignificante y no afectó a la cuantificación de dichos compuestos en las matrices de langostino y trucha.
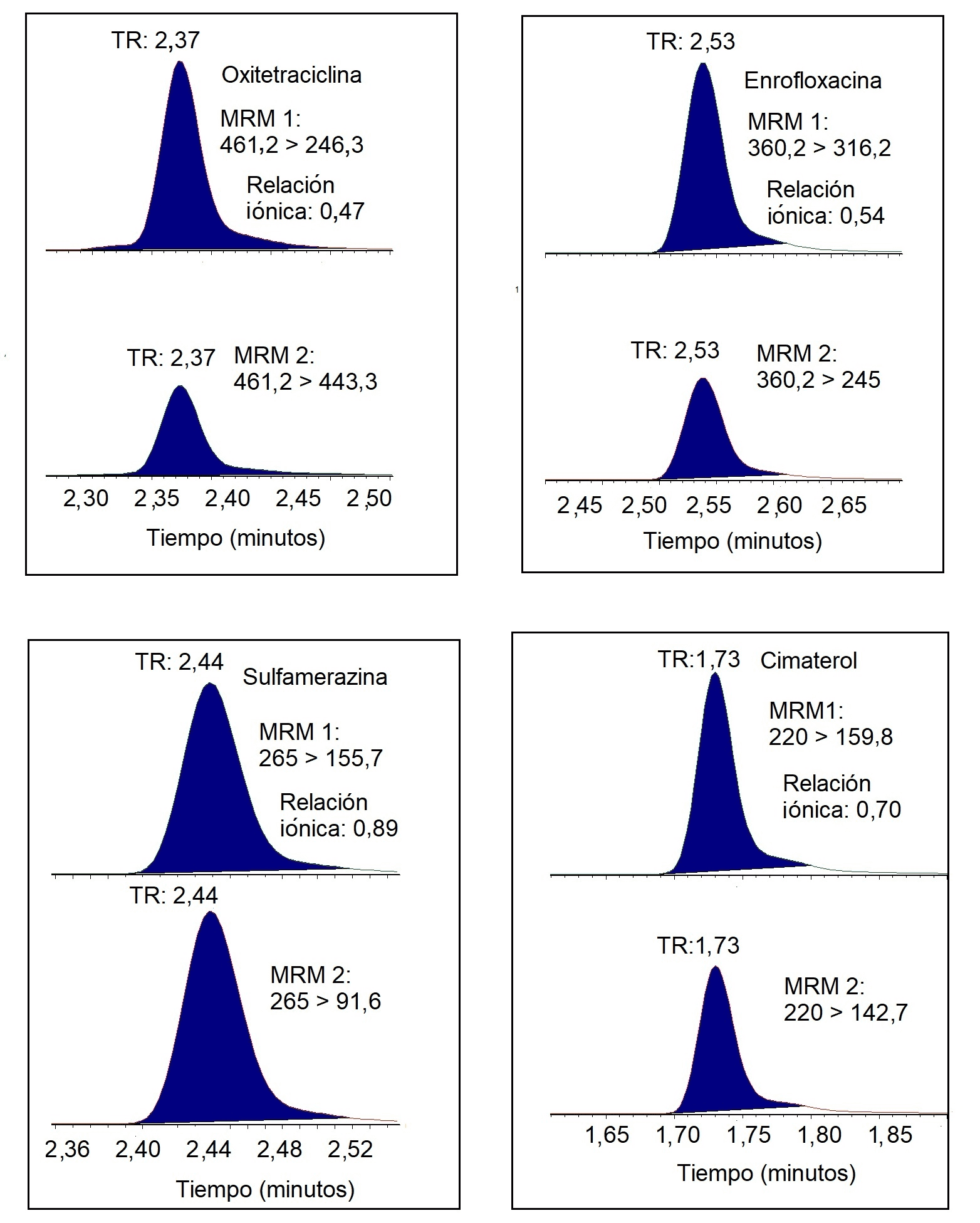
Figura 4.
Figura 4. Cromatogramas MRM de compuestos representativos fortificado a 100 µg/kg en blanco de matriz de trucha. TR: tiempo de retención; MRM: monitoreo de reacción múltiple.
Incertidumbre
La incertidumbre expandida de los medicamentos veterinarios incluidos en el estudio de validación varió desde 8% a 59% para trucha y langostino, con excepción del compuesto sulfaquinoxalina en langostino, para el que se obtuvo un valor de 98%, debido principalmente a la alta contribución de la incertidumbre de la curva de calibración, tal como se observa en la Figura 5, lo que se originó probablemente debido a errores aleatorios o sistemáticos en la preparación de la curva de calibración.
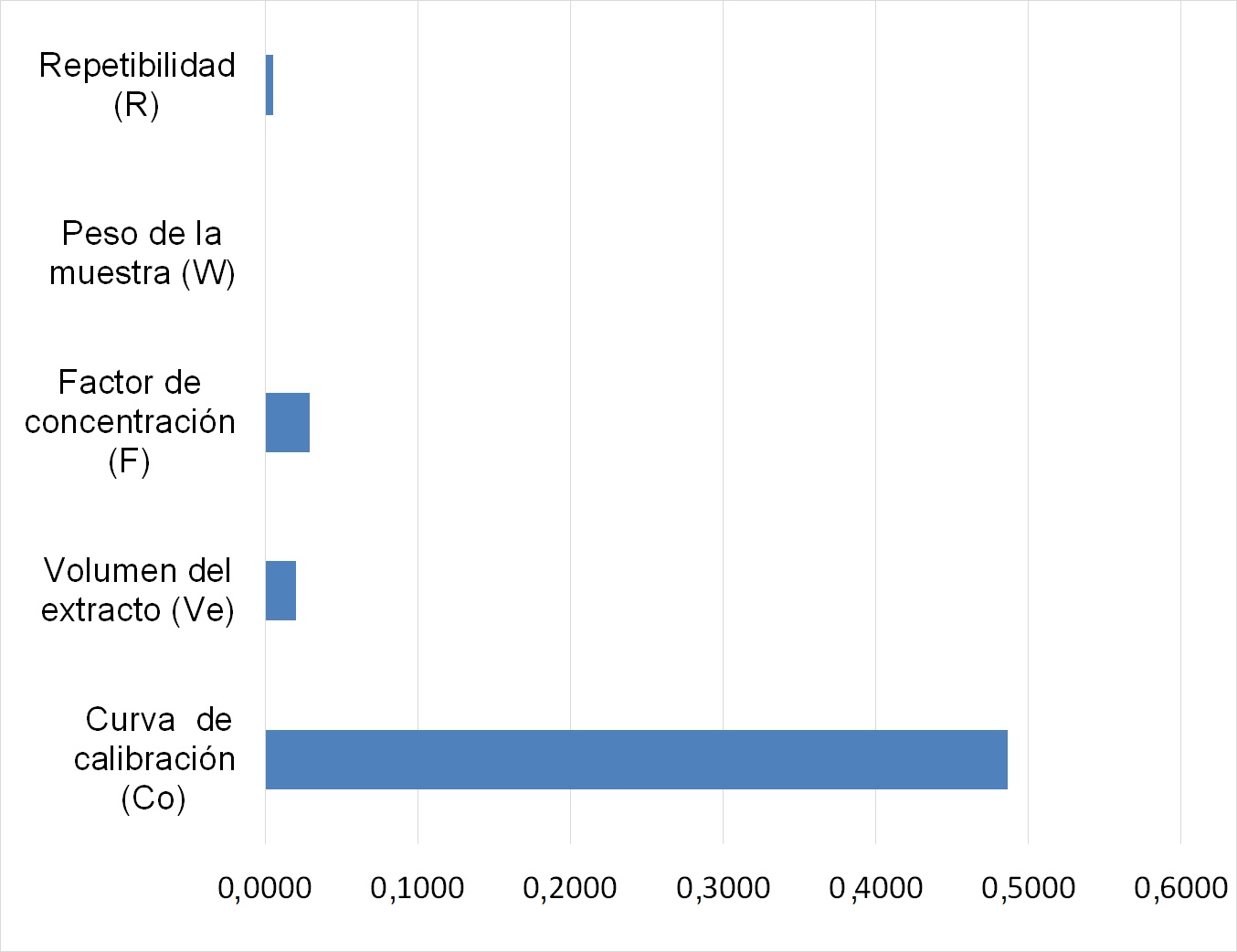
Figura 5
Figura 5. Fuentes de incertidumbre estándar relativa de sulfaquinoxalina en langostino a 50 µg/kg.
Análisis en muestras reales
Una vez desarrollado el método, fue aplicado para la determinación de residuos de medicamentos veterinarios en 6 muestras de trucha, las cuales fueron recolectadas en 6 supermercados diferentes del Cercado de Lima, Perú. Con el fin de asegurar la calidad de los resultados, se fortificó una muestra blanco a la concentración de 50 µg/kg antes de comenzar con la extracción. Además, el tiempo de retención, los fragmentos de cuantificación y confirmación y el ratio iónico encontrados en las muestras de mercado fueron comparados con los de los estándares de calibración, para su correcta identificación y cuantificación, tal como lo establece la Decisión de la Comisión 657/2002/CE [11]. Se reportaron trazas del medicamento oxitretraciclina en una de las 6 muestras, la cual se encontraba muy por debajo del LMR de 100 µg/kg. Dichos resultados concuerdan con otros trabajos de investigación reportados previamente utilizando la misma técnica analítica, en los que se detectaron pocos analitos en muestras de trucha [18]-[21].
Conclusiones
El estudio cumplió con el objetivo de desarrollar y validar un método de análisis sencillo, robusto y efectivo para la determinación multirresiduo de múltiples medicamentos veterinarios pertenecientes a diferentes familias de fármacos, en trucha y langostino. El método, que utiliza dSPE con C18 y detección por cromatografía líquida acoplada a espectrometría de masas, cumplió con los criterios de validación establecidos por el Codex Alimentarius. Los resultados obtenidos nos permiten recomendar la aplicación del método en los programas nacionales de monitoreo de la inocuidad de truchas y langostinos provenientes de acuicultura.
Agradecimientos
Los autores agradecen al Programa Nacional de Innovación en Pesca y Acuicultura (PNIPA) y a la Universidad Peruana Cayetano Heredia (UPCH) por el financiamiento y por el apoyo durante el desarrollo de la investigación. Al CITEacuícola UPCH por haber facilitado los recursos humanos, la infraestructura y los equipos de su Laboratorio de Control de Calidad y Seguridad Alimentaria para la realización del estudio.
Referencias
[1] Fondo de las Naciones para la Alimentación y la Agricultura, “El estado mundial de la pesca y la acuicultura 2020. La sostenibilidad en acción”, 2020.
https://www.fao.org/3/ca9229es/ca9229es.pdf. [Último acceso: 07 03 2022].
[2] P. van der Sleen et al., “Interannual temperature variability is a principal driver of low-frequency fluctuations in marine fish populations”, Commun. Biol., vol. 5, no. 1, pp. 1-8, 2022. DOI: 10.1038/s42003-021-02960-y
[3] ComexPerú-Sociedad de Comercio Exterior del Perú, “Exportaciones acuícolas crecieron un 25,3% en el período enero-octubre de 2021”, 2022. https://www.comexperu.org.pe/articulo/exportaciones-acuicolas-crecieron-un-253-en-el-periodo-enero-octubre-de-2021. (Último acceso: 08 03 2022).
[4] D. Schar, E. Klein, R. Laxminarayan, M. Gilbert, and T. Van Boeckel, “Global trends in antimicrobial use in aquaculture”, Sci. Rep., vol. 10, no. 1, pp. 1-9, 2020. DOI:10.1038/s41598-020-78849-3.
[5] F. M. Treiber and H. Beranek-Knauer, “Antimicrobial residues in food from animal origin—A review of the literature focusing on products collected in stores and markets worldwide”, Antibiotics, vol. 10, no. 5, pp. 1-15, 2021. DOI: 10.3390/antibiotics10050534.
[6] D. Schar, C. Zhao, Y. Wang, D. G. J. Larsson, M. Gilbert, and T. P. Van Boeckel, “Twenty-year trends in antimicrobial resistance from aquaculture and fisheries in Asia”, Nat. Commun., vol. 12, no. 1, pp. 1-10, 2021. DOI: 10.1038/s41467-021-25655-8. PMID: 34508079.
[7] Administrative Committee of the Federal Register, “Electronic Code of Federal Regulations. Title 21 Food and Drugs. Part 556. Tolerances For Residues Of New Animal Drugs In Food”, 2022. https://www.ecfr.gov/cgi-bin/text-idx?SID=45a74e7727c3e71d2325986fd8064238&mc=true&tpl=/ecfrbrowse/Title21/21cfr556_main_02.tpl. [Último acceso: 15 02 2022].
[8] H. N. Jung, D. H. Park, J. Y. Choi, S. H. Kang, H. J. Cho, J. M. Choi, J. H. Shim, A. A. Zaky, A. M. Abd El-Aty, and H. C. Shin, “Simultaneous Quantification of Chloramphenicol, Thiamphenicol, Florfenicol, and Florfenicol Amine in Animal and Aquaculture Products Using Liquid Chromatography-Tandem Mass Spectrometry”, Front. Nutr., vol. 8, no. 812803, pp. 1-10, 2022. DOI: 10.3389/fnut.2021.812803.
[9] T. J. Centner and L. Petetin, “Divergent Approaches Regulating Beta Agonists and Cloning of Animals for Food: USA and European Union”, Society & Animals, vol. 28, no. 5-6 pp. 613-32, 2020. DOI:10.1163/15685306-12341567.
[10] Fondo de las Naciones para la Alimentación y la Agricultura-Organización Mundial de la Salud, “Codex Alimentarius. CX/MRL 2-2018. Límites Máximos de Residuos (LMR) y recomendaciones sobre la gestión de riesgos (RGR) para residuos de medicamentos veterinarios en los alimentos”, 2018. https://www.fao.org/fao-who-codexalimentarius/sh-proxy/es/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXM%2B2%252FMRL2s.pdf. [Último acceso: 15 02 2022].
[11] Official Journal of the European Union. “Commission Implementing Regulation (EU) 2021/808 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to be used for sampling and repealing Decisions 2002/657/EC and 98/179/EC”, 2021. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32021R0808. [Último acceso: 08 03 2022].
[12] FDA Foods Program Regulatory Science Steering Committee, “Guidelines for the Validation of Chemical Methods in Food, Feed, Cosmetics, and Veterinary Products”, Fda.gov, 2019. https://www.fda.gov/media/81810/download. [Ultimo acceso: 08 03 2022].
[13] Fondo de las Naciones para la Alimentación y la Agricultura-Organización Mundial de la Salud, “CAC/GL 71-2009. Adoptadas en 2009. Revisadas en 2012, 2014. Directrices para el diseño y la implementación de programas nacionales reglamentarios de aseguramiento de inocuidad alimentaria relacionados con el uso de medicamentos veterinarios en los animales destinados a la producción de alimentos”, 2014. https://www.fao.org/fileadmin/user_upload/livestockgov/documents/CXG_071s.pdf. [Último acceso: 15 02 2022].
[14] S. Dugheri, N. Mucci, G. Cappelli, L. Trevisani, A. Bonari, E. Bucaletti, D. Squillaci, and G. Arcangeli, “Advanced Solid-Phase Microextraction Techniques and Related Automation: A Review of Commercially Available Technologies”, J. Anal. Methods Chem., vol. 2022, pp.1-15, 2022. DOI: 10.1155/2022/8690569.
[15] B. Magnusson and U. Örnemark, “The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics”, 2014. ISBN 978-91-87461-59-0. www.eurachem.org. [Último acceso: 15 02 2022].
[16] International Organization for Standardization / International Electrotechnical Commission, “ISO/IEC GUIDE 98-3:2008. Uncertainty of measurement — Part 3: Guide to the expression of uncertainty in measurement (GUM:1995)”, 1995. https://isotc.iso.org/livelink/livelink/fetch/2000/2122/4230450/8389141/ISO_IEC_Guide_98%2D3_2008%28E%29_%2D_Uncertainty_of_measurement_%2D%2D_Part_3%2C_Guide_to_the_expression_of_uncertainty_in_measurement_%28GUM%2C1995%29.pdf?nodeid=8389142&vernum=-2. [Último acceso: 15 02 2022].
[17] S. L. R. Ellison and A. Williams, “EURACHEM/CITAC Guide. Quantifying Uncertainty in Analytical Measurement”, 2012. https://www.eurachem.org/images/stories/Guides/pdf/QUAM2012_P1.pdf. [Último acceso: 15 02 2022].
[18] C. Cháfer-Pericás, Á. Maquieira, R. Puchades, B. Company, J. Miralles, and A. Moreno, “Multiresidue determination of antibiotics in feed and fish samples for food safety evaluation. Comparison of immunoassay vs LC-MS-MS”, Aquac. Res., vol. 22, no. 6, pp. 993-99, 2011. DOI: 10.1016/j.foodcont.2010.12.008.
[19] A. Juan-García, G. Font, and Y. Picó, “Simultaneous determination of different classes of antibiotics in fish and livestock by CE-MS”, Electrophoresis, vol. 28, no. 22, pp. 4180-91, 2007. DOI: 10.1002/elps.200700383.
[20] M. E. Dasenaki and N. S. Thomaidis, “Multi-residue determination of seventeen sulfonamides and five tetracyclines in fish tissue using a multi-stage LC-ESI-MS/MS approach based on advanced mass spectrometric techniques”, Anal. Chim. Acta, vol. 672, no. 1-2, pp. 93-102, 2010. DOI: 10.1016/j.aca.2010.04.034. Epub 2010 Apr 24. PMID: 20579496.
[21] R. Pereira Lopes, R. Cazorla Reyes, R. Romero-González, J. L. Martínez Vidal, and A. Garrido Frenich, “Multiresidue determination of veterinary drugs in aquaculture fish samples by ultra-high performance liquid chromatography coupled to tandem mass spectrometry”, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., vol. 895-896, pp. 39-47, 2012. DOI: 10.1016/j.jchromb.2012.03.011.
[22] B. Toussaint, M. Chedin, G. Bordin, and A. R. Rodriguez, “Determination of (fluoro)quinolone antibiotic residues in pig kidney using liquid chromatography–tandem mass spectrometry: I. Laboratory-validated method”, J. Chromatogr. A., vol. 1088, no. 1-2, pp. 32-39, 2005. DOI: 10.1016/j.chroma.2005.02.057.
[23] M. P. Hermo, D. Barron, and J. Barbosa, “Determination of multiresidue quinolones regulated by the European Union in pig liver samples. High-resolution time-of-flight mass spectrometry versus tandem mass spectrometry detection”, J. Chromatogr. A., vol. 1201, no. 1, pp. 1-14, 2008. DOI: 10.1016/j.chroma.2008.05.090.
[24] G. van Vyncht, A. Janosi, G. Bordin, B. Toussaint, G. Maghuin-Rogister, E. De Pauw, and A. R. Rodriguez, “Multiresidue determination of (fluoro)quinolone antibiotics in swine kidney using liquid chromatography– tandem mass spectrometry”, J. Chromatogr. A., vol. 952, no. 1-2, pp.121-29, 2002. DOI: 10.1016/s0021-9673(02)00092-4.
[25] P. Mottier, Y. A. Hammel, E. Gremaud, and P. A. Guy, “Quantitative high-throughput analysis of 16 (fluoro)quinolones in honey using automated extraction by turbulent flow chromatography coupled to liquid chromatograph- tandem mass spectrometry”, J. Agric. Food Chem., vol. 56, no. 1, pp. 35-43, 2008. DOI: 10.1021/jf072934d.
[26] S. Bogialli, G. D’Ascenzo, A. Di Corcia, A. Lagana, and G. Tramontana, “Simple assay for monitoring seven quinolone antibacterials in eggs: Extraction with hot water and liquid chromatography coupled to tandem mass spectrometry Laboratory validation in line with the European Union Commission Decision 657/2002/EC”, J. Chromatogr. A., vol. 1216, no. 5, pp. 794-780, 2009. DOI: 10.1016/j.chroma.2008.11.070.
Notas de autor
maria.rivera.c@upch.pe
Recibido: 16 de febrero de 2022; Revisión solicitada: 8 de agosto de 2022; Aceptado: 18 de agosto de 2022
Resumen
Este estudio se realizó con el objetivo de desarrollar y validar un método para la determinación de 30 medicamentos veterinarios en muestras de trucha y langostino. El método utiliza extracción en fase sólida dispersiva (dSPE) con C18 y detección por cromatografía líquida acoplada a espectrometría de masas. Se determinó linealidad, veracidad (porcentaje de recuperación), repetitividad y reproducibilidad intralaboratorio (porcentaje de desviación estándar relativa (% RSD)), límites de detección (LoD), límites de cuantificación (LoQ), selectividad e incertidumbre. La recuperación varió de 70 a 120% y la repetibilidad y la reproducibilidad fueron menores de 20% de la desviación estándar relativa. La selectividad fue adecuada, sin picos interferentes. Las relaciones iónicas cumplieron con los criterios de confirmación. Los coeficientes de determinación (R2) fueron mayores de 0,99, con excepción de la sulfaquinoxalina en langostino (R2 = 0,97). Los LoD y los LoQ variaron entre 0,6 µg/kg y 12,8 µg/kg y los valores de incertidumbre entre 6 µg/kg y 49 µg/kg. Se analizaron adicionalmente 6 muestras de diferentes mercados de Lima y se detectaron trazas de algunos medicamentos incluidos en el ensayo. El método es adecuado para el análisis de residuos de medicamentos veterinarios y se recomienda su aplicación en los programas nacionales de monitoreo de la inocuidad de truchas y langostinos provenientes de acuicultura.
Palabras clave
multirresiduos, residuos de medicamentos veterinarios, UPLC-MS, MS, truchas, langostinos.Abstract
This study was aimed at developing and validate an analysis method to determine residues of 30 veterinary drugs in rainbow trout and shrimp specimens. The method involves extraction in dispersive solid phase with C18 and the subsequent detection through liquid chromatography coupled to mass spectrometry. Validation was done through determination of linearity, trueness (% of recovery), repeatability and intralaboratory reproducibility, limits of detection (LoD), limits of quantification (LoQ) selectivity and uncertainty. Recovery ranged from 70 to 120% and repeatability and intralaboratory reproducibility were lower than 20%. Selectivity was adequate, without interference peaks. Likewise, the ionic relationships met the confirmation criteria. The linearity was adequate, with determination coefficients (R2) above 0.99, except for sulfaquinolaxin in shrimp specimens (R2 = 0,97). LoD and LoQ varied from 0,6 µg/kg to 12,8 µg/kg. Limits of uncertainty ranged from 6 µg/kg to 49 µg/kg. The method was used to analyze 6 samples from different markets in Lima (Peru), identifying traces of some drugs included in the study. Our results show that the method is adequate for the analysis of veterinary drug residues and allow us to recommend its application in national monitoring programs, to assess the safety of rainbow trout and shrimp specimens from aquaculture.
Keywords
Veterinary drug residues, UPLC, MS, MS, rainbow trouts, shrimps.Resumo
O estudo foi realizado com o objetivo de desenvolver e validar um método para a determinação de 30 medicamentos veterinários, em amostras de truta e camarão. O método utiliza extração dispersiva em fase sólida com C18 e detecção por cromatografia líquida acoplada à espectrometria de massas. Foram determinados a linearidade, a veracidade (recuperação percentual), a repetibilidade, a reprodutibilidade intra-laboratorial, os limites de detecção (LoD) e de quantificação (LoQ), a linearidade, a selectividade e a incerteza. A recuperação variou de 70 a 120%, a repetibilidade e reprodutibilidade estiveram abaixo do 20% do desvio padrão relativo. A selectividade fio adequada, sem picos de interferentes. As proporções de íons atenderam aos critérios de confirmação. Os coeficientes de determinação (R2) foram superiores a 0,99, com excepção da sulfanoxalina em camarão (R2 = 0,97). LoD e LoQ variavam entre 0,6 µg/kg e 12,8 µg/kg e valores de incerteza entre 6 µg/kg e 49 µg/kg. Seis amostras de mercados do Lima foram adicionalmente analisadas e foram detectados vestígios de alguns medicamentos incluídos no estudo. O método é adequado para o análise de resíduos de medicamentos veterinários e sua aplicação é recomendada em programas nacionais de controlo da segurança da truta e do camarão provenientes da aquicultura.
Palavras-chave
multi-resíduos, resíduos de medicamentos veterinários, UPLC-MS, MS, truta, camarão.Introducción
La acuicultura mundial muestra un incremento anual sostenido, según los reportes de la Organización de las Naciones Unidas para la Alimentación y la Agricultura [1]. La magnitud de extracción de productos provenientes de la pesca es oscilante, con épocas de mayor o menor biomasa marina extraída, en tanto, la producción proveniente de la acuicultura es más predecible, puesto que es controlada y el proceso de cría y su rendimiento se pueden modificar a voluntad [2]. En el Perú, las exportaciones acuícolas crecieron un 25,3% en el periodo enero-octubre de 2021, hasta alcanzar 338 millones de dólares estadounidenses y 43.691 toneladas [3].
Sin embargo, los productos acuícolas pueden contener residuos o contaminantes químicos, tales como residuos de medicamentos veterinarios, resultantes del uso intencionado (administración de fármacos para contrarrestar enfermedades) o no intencionado (contaminación ambiental), lo que genera un problema de salud pública y al mismo tiempo pone en riesgo la comercialización de los productos acuícolas, sobre todo en el mercado internacional [3], [4].
Medicamentos veterinarios tales como antibióticos y antiparasitarios cumplen un rol importante en la acuicultura para el tratamiento y prevención de enfermedades transmisibles. Sin embargo, cuando su uso es indiscriminado, cuando no se cumplen los periodos de retiro y las dosis establecidas, o cuando se utilizan sustancias no autorizadas, se pueden originar residuos de estas sustancias en los tejidos comestibles, que pueden causar efectos adversos en la salud humana, desde leves reacciones alérgicas hasta resistencia bacteriana y graves efectos cancerígenos o teratógenos [5], [6]. Por ello, las agencias gubernamentales de inocuidad alimentaria y en el ámbito internacional agencias como el Codex Alimentarius han establecido las concentraciones o límites máximos de residuos (LMR) que debe contener un alimento para ser considerado inocuo para el consumo humano [7]-[10]. Los exportadores de productos acuícolas deben cumplir con los requisitos sanitarios de LMR, establecidos por las autoridades de los países de destino, para evitar posibles detenciones y rechazos de sus envíos debido a la presencia de residuos o contaminantes en cantidades superiores a las permitidas. Los LMR de los medicamentos veterinarios incluidos en el presente estudio varían desde 100 hasta 2000 µg/kg; asimismo, el cloranfenicol y los beta-agonistas se encuentran prohibidos para uso en la producción animal, según lo establecido por el Código de Regulación de los Estados Unidos [7], por la Regulación Europea [8], [9] y por el Codex Alimentarius [10].
Los laboratorios que realizan ensayos de residuos de medicamentos veterinarios juegan un rol importante en el control de la inocuidad alimentaria de productos de acuicultura en el mercado nacional e internacional. En el Perú, la entidad encargada de realizar los controles oficiales de residuos de medicamentos veterinarios en los productos acuícolas es el Organismo Nacional de Sanidad Pesquera, que realiza muestreos en centros de producción y en lotes de exportación, y envía las muestras a laboratorios extranjeros para su análisis, debido a la no disponibilidad de laboratorios nacionales que realicen estos ensayos.
La cromatografía líquida acoplada a espectrometría de masas en tándem (UPLC-MS/MS) es la técnica de referencia recomendada por la Unión Europea [11], [12] y por la guía de validación de la administración de alimentos y medicamentos de los Estados Unidos [13] para realizar los análisis de residuos de medicamentos veterinarios en los alimentos, debido a su alta sensibilidad y selectividad, que permiten realizar el estudio simultáneo de diferentes grupos de fármacos y/o sus metabolitos en niveles de trazas, en el orden de partes por billón y en concentraciones menores de los LMR. La UPLC-MS/MS genera información de la estructura molecular de los analitos, a partir del monitoreo de los iones productos originados por la disociación de la molécula precursora, que se utiliza para confirmar la identidad de los analitos [11]–[13]. Esta característica de la UPLC-MS/MS ayuda a evitar o disminuir la ocurrencia de resultados falsos positivos o falsos negativos, que pueden originar problemas de detenciones o rechazos de los envíos, así como pérdidas económicas y disputas legales en el comercio internacional de alimentos. Además, la UPLC-MS/MS permite realizar análisis multirresiduo para la detección y cuantificación simultánea de cientos de moléculas en una misma corrida, sin necesidad de una rigurosa separación cromatográfica, lo cual simplifica los procesos analíticos y facilita una rápida respuesta de los laboratorios de ensayo en el control de la inocuidad alimentaria [11].
La interferencia de matriz es una de las principales desventajas de la técnica UPLC-MS/MS, que se origina por la presencia de ciertos compuestos orgánicos de los alimentos que permanecen como impurezas en los extractos de las muestras y afectan la ionización de los analitos. Para superar esta interferencia se requiere realizar una exhaustiva limpieza de los extractos, mediante uno o más métodos de purificación, tales como la extracción en fase sólida, extracción líquido-líquido, extracción asistida por microonda o extracción acelerada por solventes. Sin embargo, los laboratorios que realizan análisis rutinarios requieren utilizar métodos sencillos con la finalidad de generar resultados rápidos para el control de la inocuidad alimentaria. La dSPE, introducida para el análisis de residuos de plaguicidas en frutos y vegetales [14], es actualmente la técnica de extracción y limpieza de muestras de mayor aceptación internacional en los laboratorios que realizan análisis para el control de la inocuidad alimentaria.
El presente estudio se realizó con el objetivo de desarrollar y validar un método de análisis sencillo, robusto y efectivo para la determinación multirresiduo de 30 medicamentos veterinarios pertenecientes a diferentes familias de fármacos (penicilinas, tetraciclinas, macrolidos, entre otros), en trucha (Oncorhynchus mykiss) y langostino (Litopenaeus vannamei). El método presentado en este artículo utiliza dSPE con C18 y un posterior análisis instrumental por UPLC-MS/MS.
Materiales y métodos
Estándares y reactivos
Para evaluar los medicamentos veterinarios de interés se utilizaron estándares de alta pureza (mayor de 99%). Los estándares de tetraciclina, clortetraciclina, oxitetraciclina, ácido oxolínico, flumequina, ácido nalidíxico, enrofloxacino, ciprofloxacino, sulfamerazina, sulfacloropiridazina, sulfadiazina, sulfametoxazol, sulfaquinolaxina, sulfametazina, sulfatiazol, sulfadimetoxina, sulfametoxipiridazina, sulfadoxina, espiramicina, eritromicina, trimetoprim, cimaterol, salbutamol, Terbutalina, ractopamina, zilpaterol, clembuterol, florfenicon y cloranfenicol fueron adquiridos de Sigma Aldrich (San Luis, EE. UU.). Los estándares de amoxicilina y mapenterol fueron adquiridos de Dr. Ehrenstorfer (Augsburg, Alemania).
Los reactivos utilizados fueron de grado LC-MS o HPLC. Metanol y acetonitrilo fueron obtenidos de Sigma-Aldrich, ácido fórmico de Fluka, sulfato de magnesio anhidro y acetato de sodio anhidro de JT Baker (Deventer, Holland) y agua ultrapura del equipo Milipore Mili-Q system (Milford, MA, EE. UU.).
Las soluciones individuales concentradas de 2000 ng/µL (soluciones stock) de cada estándar de medicamento fueron preparadas disolviendo una cantidad apropiada de cada estándar primario en acetonitrilo, metanol o agua ultrapura. Las soluciones fueron almacenadas a -20 °C en la oscuridad. Las soluciones de mezclas estándares de 40 ng/µL fueron preparadas tomando una alícuota de cada una de las soluciones stock y disolviéndolas en acetonitrilo, con excepción de los betalactámicos, en los cuales se utilizó agua para la disolución y almacenamiento en viales plásticos, a fin de evitar problemas de absorción en vidrio de los antibióticos. Las soluciones de calibración de 10 ng/µL y 2 ng/µL fueron preparadas disolviendo en acetonitrilo una cantidad apropiada de las soluciones de mezclas de estándares de 40 ng/µL.
Preparación de las muestras
Las muestras de truchas y langostinos fueron obtenidas de empresas locales dedicadas a la acuicultura. Fueron mantenidas en almacenamiento a -20 °C y protegidas de la luz hasta su preparación para el análisis. Así mismo, fueron descongeladas y llevadas a temperatura ambiente antes de realizar los ensayos. Se procedió a la molienda de langostinos (sin cabeza y antenas) y trucha (solo músculo) utilizando el homogenizador Robot coupe Blixer 3.0. Luego se realizó la extracción de la siguiente manera: se pesaron aproximadamente 2,00 g de músculo de trucha o langostino dentro de tubos de centrifuga de polipropileno de 50 mL, luego se agregaron 10 mL de acetonitrilo/EDTA 0.1 M en agua (4/1%v/v) y se homogeneizó con un equipo vortex por 5 min, luego se procedió a centrifugar por 5 min a 4000 rpm, el sobrenadante se trasvasó a un tubo de 50 mL que contenía 300 mg de adsorbente C18; luego se adicionaron 10 mL de hexano presaturado en acetonitrilo, se agitó por 30 s y se procedió a centrifugar nuevamente por 5 min a 4000 rpm. Se eliminó la capa de hexano con una pipeta Pasteur, del sobrenadante que quedó se tomó una alícuota de 5 mL y se llevó a un tubo de centrifuga de polipropileno de 15 mL. Finalmente, se concentró el extracto a sequedad, en un equipo de evaporación de nitrógeno a 45 °C. El extracto evaporado fue reconstituido con 1 mL de fase móvil A (ácido fórmico al 0,1% en agua), se filtró a través de filtros de difluoruro de polivinilo (PDVF) de 0,2 µm y se colocó en viales de color ámbar. Finalmente, se inyectaron 2 µL en el equipo de UPLC-MS/MS.
Análisis por UPLC-MS/MS
El análisis por cromatografía se realizó con el equipo Acquity UPLC system (Waters, Milford, MA, EE. UU.) y la separación de los analitos se realizó con la columna Acquity UPLC BEH C18 (100 mm x 2,1 mm, 1,7 µm). Para la corrida cromatográfica se utilizó como fase móvil ácido fórmico al 0,1% en agua ultrapura (Fase A) y 0,1% ácido fórmico en acetonitrilo (Fase B) con un flujo de 0,5 mL/min. El gradiente de elución inició con 0% fase móvil B por 0,1 min, que se incrementó en forma lineal hasta 100% en 7,9 min, y se mantuvo por 1,5 min antes de regresar a la condición inicial en 0,1 min, con un tiempo de preequilibración de 3,4 min. El tiempo de la corrida cromatográfica fue de 13 min, el volumen de inyección fue de 2 µL y la temperatura de la columna fue de 40 °C.
El análisis por espectrometría de masas se realizó con el equipo UPLC-MS/MS Acquity Xevo TQ-XS (Waters, Manchester, Reino Unido). El equipo fue operado utilizando la fuente de ionización por electrospray (ESI) en modo ESI positivo. Los parámetros para la fuente de ionización fueron: voltaje de capilar 3,0 kV, temperatura de la fuente 150 °C, temperatura de desolvatación 500 °C, flujo del gas de cono 150 L/h y flujo del gas de desolvatación 1000 L/h. La colisión inducida por disociación se realizó utilizando argón como gas de colisión, a una presión 4 x10-3
mbar en la celda de colisión. La adquisición y procesamiento de datos en el equipo UPLC-MS/MS se realizó con el programa MassLynx 4.2. En la Tabla 1 se muestran los principales parámetros de espectrometría de masa MS/MS por cada analito determinado.
aIon producto más abundante utilizado para el análisis cuantitativo.
bEnergía de colisión.
cIon producto segundo en abundancia, utilizado para la detección cualitativa.Tabla 1:
Tabla 1. Ventanas de tiempo de retención tR y condiciones MSMS de los analitos incluidos en el estudio
Analito
Grupo farmacológico
Ventana tR (min)
Cono (V)
Ion precursor (m/z)
Iones productos (m/z)
Cuantia (m/z)
ECb (eV)
Cualic (m/z)
EC (eV)
1
Oxitetraciclina
Tetraciclinas
1,98 – 2,78
40
461v3
426,3
18
443,3
12
2
Clortetraciclina
Tetraciclinas
2,59 – 3,39
20
479,4
444,2
20
462,2
15
3
Tetraciclina
Tetraciclinas
2,11 – 2,91
20
445,3
410,3
18
153,7
25
4
Enrofloxacino
Quinolonas
2,14 – 2,94
52
360,2
316,2
18
245
24
5
Flumequina
Quinolonas
3,79 – 4,59
40
262
244
18
201,8
32
6
Ciprofloxacino
Quinolonas
1,98 – 2,78
50
332,3
288,1
16
244,9
22
7
Ácido nalidíxico
Quinolonas
3,66 – 4,46
6
233
214,9
14
186,8
22
8
Ácido oxolínico
Quinolonas
3,05 – 3,85
24
262
244
18
159,7
34
9
Sulfamerazina
Sulfonamidas
2,04 – 2,84
34
265
91,6
26
155,7
16
10
Sulfacloropiridazina
Sulfonamidas
2,62 – 3,42
40
285,3
155,7
12
91,5
28
11
Sulfadiazina
Sulfonamidas
1,75 – 2,55
42
251
155,7
12
91,6
22
12
Sulfametoxazol
Sulfonamidas
2,78 – 3,58
10
254
155,7
14
91,5
24
13
Sulfaquinolaxina
Sulfonamidas
3,23 – 4,03
54
301,1
155,7
14
91,5
34
14
Sulfametazina
Sulfonamidas
2,29 – 3,09
42
279,1
185,8
16
123,7
25
15
Sulfatiazol
Sulfonamidas
1,84 – 2,64
38
256
155,7
15
91,5
26
16
Sulfadimetoxina
Sulfonamidas
3,22 – 4,02
56
311,1
155,7
22
91,5
32
17
Sulfametoxipiridazina
Sulfonamidas
2,30 – 3,10
46
281
155,7
15
91,5
26
18
Sulfadoxina
Sulfonamidas
2,77 – 3,57
44
311,1
155,7
16
91,5
26
19
Cimaterol
B-agonistas
1,33 – 2,13
22
220
159,8
16
142,7
23
20
Salbutamol
B-agonistas
1,27 – 2,07
34
240,1
147,7
18
165,8
12
21
Terbutalina
B-agonistas
1,26 – 2,06
16
226
151,8
14
106,6
28
22
Ractopamina
B-agonistas
2,04 – 2,84
35
302,2
163,8
15
284,2
12
23
Zilpaterol
B-agonistas
1,26 – 2,06
20
262,1
244,1
12
184,9
23
24
Clembuterol
B-agonistas
2,33 – 3,13
8
277
202,8
14
131,7
24
25
Mapenterol
B-agonistas
2,95 – 3,75
22
325,2
236,9
14
307,1
10
26
Espiramicina
Macrólidos
2,45 – 3,25
20
422,5
173,9
20
100,6
15
27
Amoxicilina
Penicilinas
1,26 – 2,06
6
366,3
349,2
6
113,5
18
28
Florfenicol
Fenicoles
2,82 – 3,62
25
358,2
240,8
16
205,8
26
29
Trimetroprim
Trimetroprim
1,88 – 2,68
40
291,1
230
22
122,6
28
30
Cloranfenicol
Fenicoles
3,01 – 3,81
30
323,2
275
12
164,8
22
Validación del método
La validación del método se realizó con la finalidad de verificar el funcionamiento analítico del método desarrollado para el uso propuesto, es decir, el análisis de residuos de medicamentos veterinarios en productos acuícolas (truchas y langostinos). La validación se realizó de acuerdo al procedimiento de la Regulación Unión Europea 2002/657 [11] y Eurachem [15]. Para ello, se utilizó muestra de blancos de matriz de truchas y langostinos fortificados con cantidades apropiadas de la solución de estándares de trabajo para obtener tres niveles de concentraciones de 10, 50 y 100 µg/kg, con 10 análisis repetidos por cada nivel. Para determinar la reproducibilidad intralaboratorio, se realizaron los análisis en tres ocasiones diferentes, con tres analistas. La linealidad fue evaluada utilizando curva de calibración fortificada en blanco de matriz en 5 niveles de concentración de 10 µg/kg hasta 300 µg/kg. Los límites de deteción (LoD) y de cuantificación (LoQ) fueron estimados utilizando blancos de matriz fortificados a la concentración de 10 µg/kg, con 10 análisis repetidos.
La evaluación de la selectividad del método por UPLC-MS/MS se realizó mediante adquisición por espectrometría de masa en modo de monitoreo de reacción múltiple (MRM), con dos iones productos por cada compuesto. La proporción de las señales de los iones productos (relaciones iónicas) de cada compuesto se halló dividiendo las señales del ion menos abundante entre el ion más abundante. La identificación de los compuestos por UPLC-MS/MS se realizó según los requisitos de relación iónica establecidos por Codex [13].
La estimación de la incertidumbre se realizó de acuerdo al procedimiento establecido en la Guía ISO o método GUM [16] y Eurachem [17]. La estimación de la incertidumbre combinada se calculó a partir de la Ec. (1):
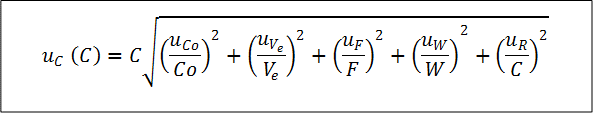
Donde C=Ccontenido en la muestra en µg/kg, Co=concentración en el extracto en ng/mL obtenida por interpolación en la curva de calibración, Ve=volumen del extracto, W=peso de la muestra, F=factor de concentración y R=repetibilidad. La incertidumbre expandida se calculó multiplicando la incertidumbre combinada por K=2, considerando un nivel de confianza de 95%.
La evaluación de los parámetros de validación se realizó de acuerdo a los criterios de aceptación establecidos por el Codex Alimentarius [13], con RSD menor de 20% para la repetibilidad y reproducibilidad intralaboratorio y con recuperación de 70% a 120% para la veracidad. La selectividad del método se evaluó comparando los cromatogramas de los blancos de matriz versus los blancos de matriz fortificados, verificando que no hubiera señales interferentes en la región en donde eluyen los analitos. Asimismo, se realizó la confirmación por espectrometría de masas comparando la relación iónica de dos fragmentos MS/MS de cada molécula, verificando que las relaciones iónicas de las muestras estuvieran dentro del intervalo de aceptabilidad, en comparación con los estándares establecidos por el Codex Alimentarius [13].
Resultados y discusión
Análisis por UPLC-MS/MS
La optimización de los parámetros de espectrometría de masa MS/MS fue realizada mediante infusión directa en el espectrómetro de soluciones de cada uno de los estándares de medicamentos veterinarios, a la concentración de 0,5 mg/L. El electrospray fue utilizado en modo ion positivo, puesto que todos los analitos presentaban mayor señal en este modo de ionización. En la Tabla 1 se muestran las condiciones del espectrómetro de masas utilizadas en el estudio. En cuanto a la condiciones cromatográficas, fueron desarrolladas principalmente para optimizar la forma de pico, resolución e intensidad de los analitos. La fase móvil fue investigada principalmente para maximizar la sensibilidad y la resolución del método. Se probaron diferentes solventes polares, tales como metanol o acetonitrilo y agua ultrapura, todos con concentraciones de ácido fórmico al 0,05 y 0,1% (v/v). El acetonitrilo presentó mayor sensibilidad para los analitos y el ácido fórmico favoreció la ionización de los analitos. De igual manera, se configuró el tiempo de monitoreo de cada transición (dwell time) utilizando el programa MassLynx 4.2, con la finalidad de obtener como mínimo 15 puntos en cada pico cromatográfico.
En la Figura 1 se muestra el cromatograma de ion total (TIC) para la matriz trucha. Los tiempos de retención de los compuestos en las matrices de trucha y langostino fueron similares; variaron desde 1,66 hasta 4,19 minutos. La gradiente utilizada sirvió para la separación de los analitos en menos de 5 minutos. A pesar de que algunos compuestos coeluyen, ello no constituyó problema para su cuantificación, debido a la alta selectividad de la técnica de UPLC-MS/MS, puesto que estos compuestos son separados y detectados mediante adquisición de monitoreo de reacción múltiple por espectrometría de masas en tándem.

Figura 1. Cromatogramas de ion total (TIC) de 30 residuos de medicamentos veterinarios, fortificados a 100 µg/kg en blanco de matriz de trucha.
Validación del método
Después de determinar las condiciones de extracción, de separación cromatográfica y de detección por espectrometría de masas, se validó el método de análisis, determinándose los parámetros de recuperación, repetitividad, reproducibilidad intralaboratorio, linealidad, LoD, LoQ, selectividad e incertidumbre.
Recuperación
En las Tablas 2 y 3 se observa que los porcentajes de recuperación variaron desde 76,8 hasta 120,5%, valores que se encuentran dentro de la especificación establecida por el Codex Alimentarius, es decir, entre 70 y 120%.
aRecuperación de 10 análisis en 03 ocasiones (n=30) (%).
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).Tabla 2:
Tabla 2. Recuperación repetibilidad (%RSDr) y reproducibilidad intralaboratorio (%RSDR) obtenidos en trucha fortificada a 10 50 y 100 µg/kg.
Analito
10 µg/kg
50 µg/kg
100 µg/kg
Ra
RSDrb
RSDR
c
R
RSDr
RSDR
R
RSDr
RSDR
1
Oxitetraciclina
100
1,9
5,4
91
11,0
11,5
93
2,3
14,1
2
Clortetraciclina
104
7,1
7,0
96
4,9
4,2
98
4,0
5,5
3
Tetraciclina
98
4,0
6,0
91
12,8
12,6
93
1,8
13,8
4
Enrofloxacino
93
2,8
8,7
97
8,8
10,4
99
2,1
12,1
5
Flumequina
106
3,6
4,1
93
9,5
10,6
98
1,0
10,1
6
Ciprofloxacino
93
5,7
5,3
96
9,1
9,5
97
2,2
10,1
7
Ácido nalidíxico
95
1,5
2,2
95
8,6
10,9
99
1,1
11,6
8
Ácido oxolínico
91
3,5
3,4
98
9,9
11,2
101
1,7
11,2
9
Sulfamerazina
89
3,9
5,3
101
7,2
9,2
107
3,3
9,3
10
Sulfacloropiridazina
99
3,1
5,9
95
5,7
7,4
100
2,4
9,5
11
Sulfadiazina
100
2.8
4,8
95
12,7
12,8
99
1,8
12,5
12
Sulfametoxazol
100
2,3
3,4
96
4,0
6,4
101
2,0
7,0
13
Sulfaquinoxalina
98
3,3
5,6
92
9,1
10,9
98
1,6
10,3
14
Sulfametazina
100
2,9
3,2
90
11,2
11,2
96
1,6
11,6
15
Sulfatiazol
96
2,8
5,2
92
9,3
10,4
98
1,6
11,0
16
Sulfadimetoxina
96
3,6
6,8
89
10,7
14,0
95
1,3
15,0
17
Sulfametoxipiridazina
103
1,6
3,4
95
10,4
12,0
101
2,8
12,0
18
Sulfadoxina
103
3,0
3,5
95
5,2
7,2
99
1,9
8,5
19
Cimaterol
77
2,8
3,6
98
5,6
7,1
101
1,6
8,9
20
Salbutamol
84
1,5
3,3
108
2,7
3,0
106
1,0
4,4
21
Terbutalina
82
4,2
4,5
106
2,2
3,0
104
1,8
4,1
22
Ractopamina
88
2,8
9,9
109
2,3
3,7
109
1,1
4,1
23
Zilpaterol
93
3,1
3,5
101
4,6
4,4
100
1,6
6,1
24
Clembuterol
96
1,9
2,8
103
2,3
1,9
106
1,5
2,7
25
Mapenterol
92
3,2
4,1
99
2,3
2,8
103
1,4
5,0
26
Espiramicina
100
3,3
3,3
98
2,8
3,0
103
2,7
4,8
27
Amoxicilina
103
3,5
4,4
95
4,3
3,7
96
2,1
6,4
28
Florfenicol
99
5,8
7,2
97
6,2
6,3
100
1,9
8,0
29
Trimetoprim
89
2,1
4,9
111
2,5
2,5
110
1,4
3,9
30
Cloranfenicol
90
6,4
7,1
94
8,4
8,1
98
3,4
11,2
aRecuperación de 10 análisis en 03 ocasiones (n=30) (%).
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).
aRecuperación de 10 análisis en 03 ocasiones (n=30) (%).
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).Tabla 3: Recuperación, repetibilidad (%RSDr) y reproducibilidad intralaboratorio (%RSDR) obtenidos en langostino fortificado a 10, 50 y 100 µg/kg.
Analito
10 µg/kg
50 µg/kg
100 µg/kg
Ra
RSDrb
RSDR
c
R
RSDr
RSDR
R
RSDr
RSDR
1
Oxitetraciclina
113
2,4
4,4
80
2,2
6,3
100
1,6
8,1
2
Clortetraciclina
116
4,6
4,3
81
9,0
9,8
97
6,1
6,9
3
Tetraciclina
115
2,6
3,2
82
3,8
4,6
99
1,8
5,4
4
Enrofloxacino
113
2,1
3,4
82
2,7
5,1
97
2,0
6,2
5
Flumequina
109
4,7
6,5
79
4,7
3,7
97
1,6
3,3
6
Ciprofloxacino
109
5,1
7,1
80
4,1
5,9
96
1,9
6,5
7
Ácido nalidíxico
112
2,5
3,4
82
2,1
2,6
100
1,0
2,8
8
Ácido oxolínico
113
1,9
3,0
84
1,5
2,5
103
1,8
3,6
9
Sulfamerazina
92
2,8
5,2
99
2,1
3,5
111
1,9
4,5
10
Sulfacloropiridazina
114
2,8
4,5
82
2,2
4,2
98
1,4
5,2
11
Sulfadiazina
110
3,1
4,8
88
2,2
2,1
101
2,5
2,4
12
Sulfametoxazol
117
3,4
2,4
83
2,6
2,6
99
1,4
1,9
13
Sulfaquinoxalina
116
3,2
6,1
79
1,9
3,6
101
1,5
4,2
14
Sulfametazina
102
3,4
4,6
91
3,4
3,4
107
1,2
3,2
15
Sulfatiazol
107
2,2
2,7
85
2,0
4,1
100
2,0
2,3
16
Sulfadimetoxina
113
0,8
2,9
81
1,6
3,9
99
1,6
3,9
17
Sulfametoxipiridazina
104
3,8
3,5
89
2,4
3,4
105
3,2
3,6
18
Sulfadoxina
115
2,4
2,4
84
1,1
2,2
100
2,1
3,9
19
Cimaterol
102
2,5
3,9
89
1,1
3,2
101
1,7
3,6
20
Salbutamol
111
1,8
3,4
85
0,6
1,9
96
1,0
2,2
21
Terbutalina
109
1,7
2,2
88
1,5
3,9
97
2,4
4,1
22
Ractopamina
115
2,1
4,4
81
2,1
3,6
101
1,4
5,0
23
Zilpaterol
114
1,4
4,4
86
1,9
4,4
99
1,2
4,1
24
Clembuterol
114
1,5
2,2
83
2,3
2,7
101
1,2
3,4
25
Mapenterol
112
1,4
3,0
82
1,7
3,6
99
2,3
4,8
26
Espiramicina
112
1,5
2,8
89
2,2
3,5
90
1,7
4,5
27
Amoxicilina
118
2,1
5,1
86
2,5
6,6
86
4,0
5,8
28
Florfenicol
121
9,0
10,6
88
6,3
6,0
103
3,6
4,8
29
Trimetoprim
107
2,0
4,0
87
2,3
3,5
104
2,1
4,1
30
Cloranfenicol
114
5,0
11,1
83
4,9
6,9
99
3,6
4,9
aRecuperación de 10 análisis en 03 ocasiones (n=30) (%).
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).
Repetibilidad y reproducibilidad intralaboratorio
En las Tablas 2 y 3 se observa que la repetibilidad y la reproducibilidad intralaboratorio, expresadas como % RSD, variaron desde 0,6% hasta 12,8%, con lo que se cumple con los criterios del Codex Alimentarius que establecen un % RSD < 20%. Cabe destacar que dichos porcentajes se encuentran en línea con otros estudios, en los cuales se validó la técnica analítica en matrices como pescados y langostinos, en los que se obtuvieron % RSD superiores a 20 % [18]-[21].
Límites de detección y de cuantificación
El LoD y el LoQ hallados en trucha y langostinos variaron desde 0,6 µg/kg hasta 12,8 µg/kg. En la Figura 2 se observa que la mayoría de los LoQ de los medicamentos incluidos fueron menores de 10 µg/kg, a excepción de cloranfenicol y florfenicol en langostino, que fueron de 12,7 y 12,8 µg/kg, respectivamente. Dichos valores están muy por debajo de los LMR de estos compuestos, que varían desde 50 hasta 2000 µg/kg, a excepción del cloranfenicol y los compuestos beta-agonistas, medicamentos prohibidos para uso en animales para consumo humano y para los que no se han establecido sus respectivos LMR.
Los LoQ hallados para cloranfenicol fueron de 6,4 y 12,7 µg/kg para trucha y langostino, respectivamente, superiores al límite de performance mínimo requerido de 0,3 µµg/kg establecido para cloranfenicol por la Regulación Europea [6], lo que se explicaría por el hecho de que, al haberse utilizado un método multirresiduo, no se puede aplicar condiciones de sintonización del espectrómetro de masas, condiciones cromatográficas y condiciones de extracción especificas con la finalidad de incrementar la sensibilidad del método para esta molécula. Por ello, lo recomendable sería realizar un método individual para el análisis del cloranfenicol, con la finalidad de obtener límites analíticos menores o iguales a lo requerido. Una situación diferente se observó para el florfenicol, para el cual se obtuvo LoQ de 7,2 y 12,8 µµg/kg para trucha y langostino, respectivamente, similares al cloranfenicol, por lo que estos límites analíticos obtenidos serian adecuados para el análisis de florfenicol, debido a que están muy por debajo del LMR, que es de 1000 µg/kg. Hay que remarcar que en investigaciones previas se han reportado valores de LoD y LoQ similares o por debajo de lo hallado en el presente estudio [18]-[26].
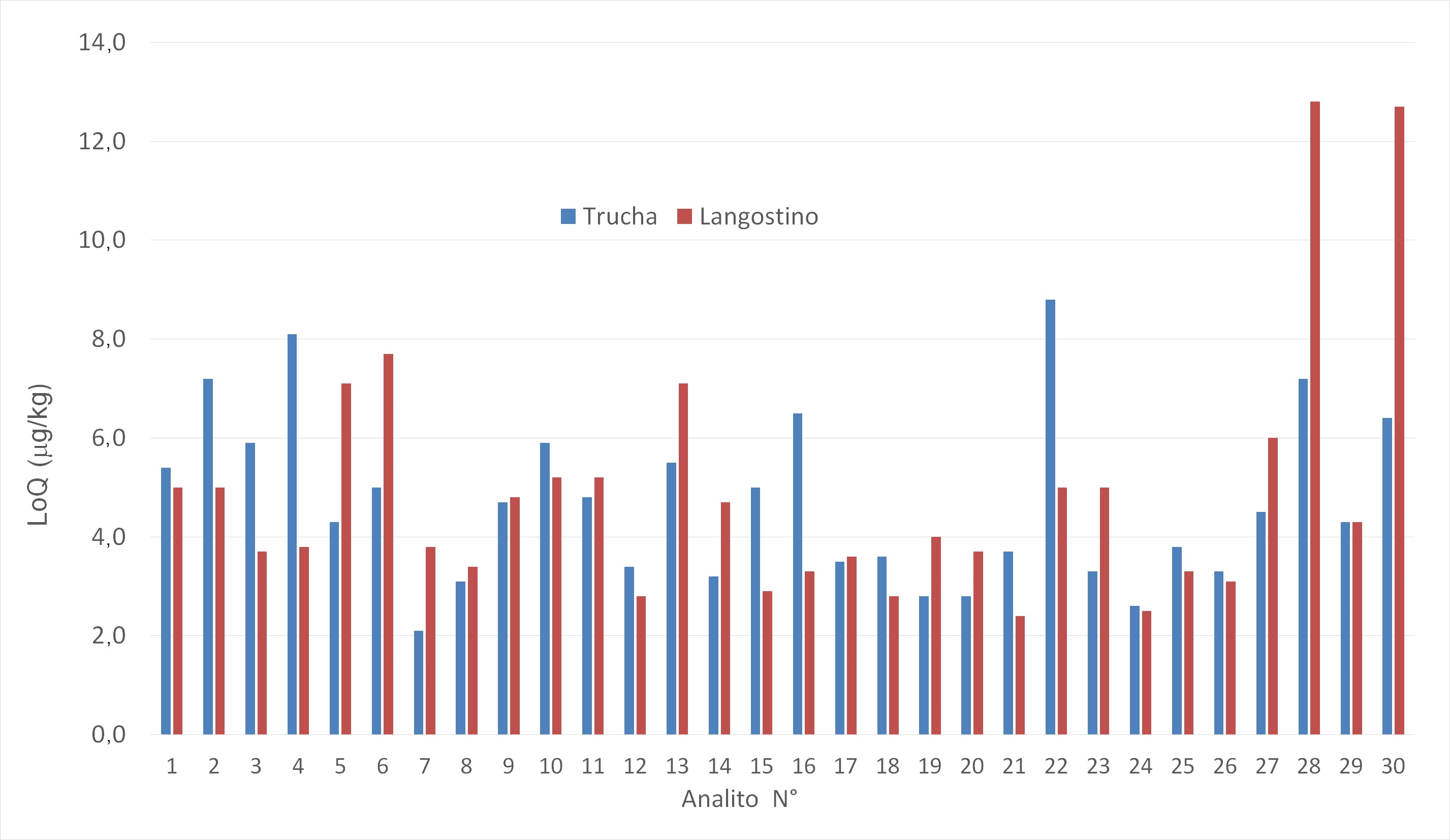
Figura 2.:
Figura 2. Límite de cuantificación (LoQ) (µg/kg) del método en trucha y langostino.
Linealidad
Los coeficientes de determinación (R.) de las curvas de calibración de los medicamentos veterinarios fortificados en trucha y langostino fueron menores de 0,99; con excepción de sulfaquinoxalina en langostino, que fue de 0,97. En la Figura 3 se observan las curvas de calibración para la matriz trucha de compuestos representativos de los principales grupos de fármacos veterinarios incluidos en el estudio de validación.
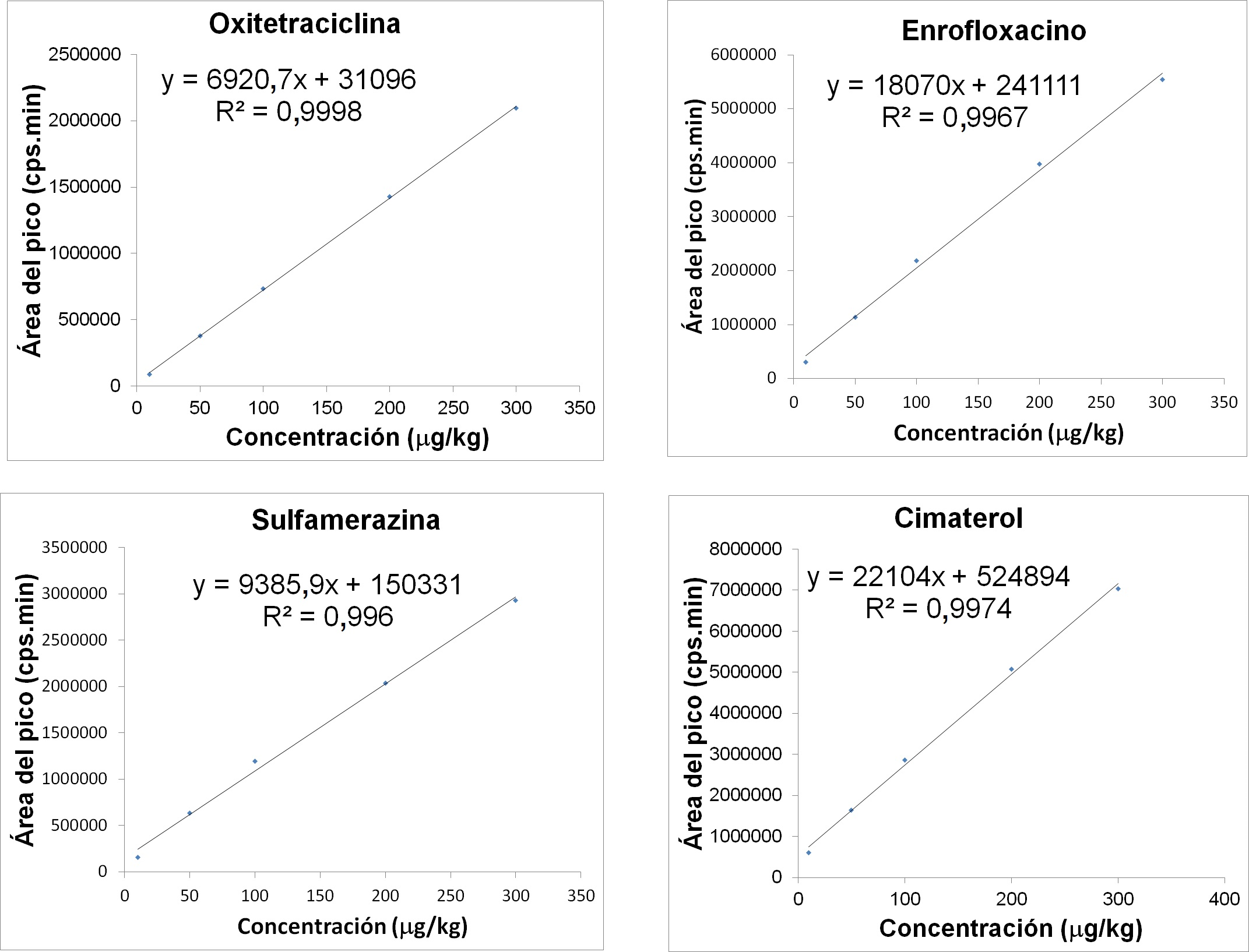
Figura 3:
Figura 3. Curva de calibración de compuestos representativos fortificados en blanco de matriz de trucha.
Selectividad
La selectividad del método se evidenció mediante identificación por LC-MSMS de los medicamentos veterinarios incluidos en el estudio, dado que las relaciones iónicas de los compuestos cumplieron con los rangos de aceptación del Codex [13]. En la Figura 4 se muestran los cromatogramas MRM de los compuestos representativos fortificados en blando de matriz trucha, en los que se observa que el monitoreo de dos iones fragmentos característicos de la estructura molecular de cada compuesto le confiere una alta selectividad a la técnica de UPLC-MS/MS. Asimismo, se identificaron las moléculas por el tiempo de retención (TR). Por otro lado, se observó efecto de arrastre en los compuestos del grupo de las quinolonas, vale decir, ácido nalidíxico, ácido oxolínico, enrofloxacina y ciprofloxacina. Para superar este efecto, se requeriría investigar los métodos de lavado del inyector utilizando diferentes solventes y tiempos de lavado. Sin embargo, debe puntualizarse que el efecto de arrastre fue insignificante y no afectó a la cuantificación de dichos compuestos en las matrices de langostino y trucha.
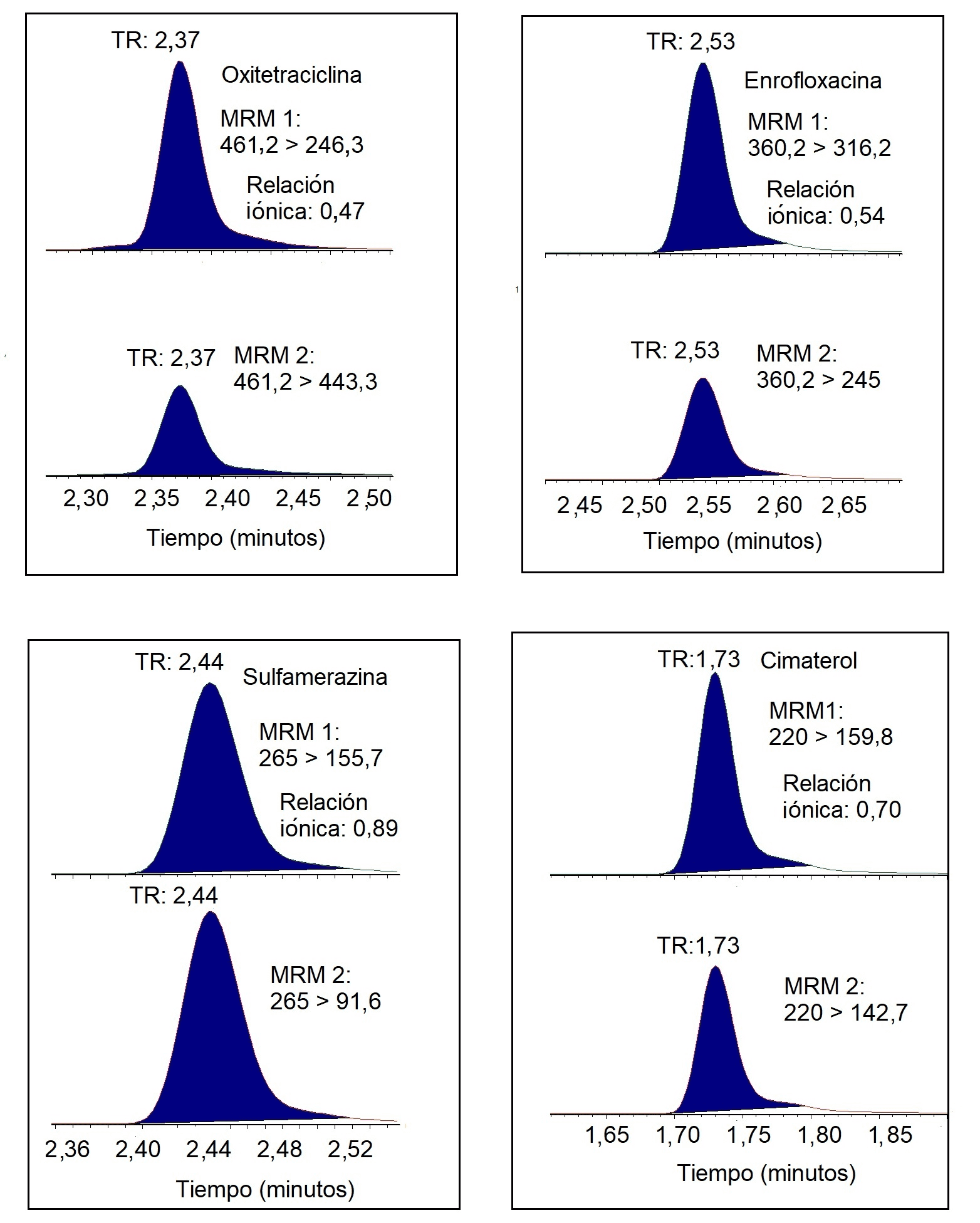
Figura 4.:
Figura 4. Cromatogramas MRM de compuestos representativos fortificado a 100 µg/kg en blanco de matriz de trucha. TR: tiempo de retención; MRM: monitoreo de reacción múltiple.
Incertidumbre
La incertidumbre expandida de los medicamentos veterinarios incluidos en el estudio de validación varió desde 8% a 59% para trucha y langostino, con excepción del compuesto sulfaquinoxalina en langostino, para el que se obtuvo un valor de 98%, debido principalmente a la alta contribución de la incertidumbre de la curva de calibración, tal como se observa en la Figura 5, lo que se originó probablemente debido a errores aleatorios o sistemáticos en la preparación de la curva de calibración.

Figura 5:
Figura 5. Fuentes de incertidumbre estándar relativa de sulfaquinoxalina en langostino a 50 µg/kg.
Análisis en muestras reales
Una vez desarrollado el método, fue aplicado para la determinación de residuos de medicamentos veterinarios en 6 muestras de trucha, las cuales fueron recolectadas en 6 supermercados diferentes del Cercado de Lima, Perú. Con el fin de asegurar la calidad de los resultados, se fortificó una muestra blanco a la concentración de 50 µg/kg antes de comenzar con la extracción. Además, el tiempo de retención, los fragmentos de cuantificación y confirmación y el ratio iónico encontrados en las muestras de mercado fueron comparados con los de los estándares de calibración, para su correcta identificación y cuantificación, tal como lo establece la Decisión de la Comisión 657/2002/CE [11]. Se reportaron trazas del medicamento oxitretraciclina en una de las 6 muestras, la cual se encontraba muy por debajo del LMR de 100 µg/kg. Dichos resultados concuerdan con otros trabajos de investigación reportados previamente utilizando la misma técnica analítica, en los que se detectaron pocos analitos en muestras de trucha [18]-[21].
Conclusiones
El estudio cumplió con el objetivo de desarrollar y validar un método de análisis sencillo, robusto y efectivo para la determinación multirresiduo de múltiples medicamentos veterinarios pertenecientes a diferentes familias de fármacos, en trucha y langostino. El método, que utiliza dSPE con C18 y detección por cromatografía líquida acoplada a espectrometría de masas, cumplió con los criterios de validación establecidos por el Codex Alimentarius. Los resultados obtenidos nos permiten recomendar la aplicación del método en los programas nacionales de monitoreo de la inocuidad de truchas y langostinos provenientes de acuicultura.
Agradecimientos
Los autores agradecen al Programa Nacional de Innovación en Pesca y Acuicultura (PNIPA) y a la Universidad Peruana Cayetano Heredia (UPCH) por el financiamiento y por el apoyo durante el desarrollo de la investigación. Al CITEacuícola UPCH por haber facilitado los recursos humanos, la infraestructura y los equipos de su Laboratorio de Control de Calidad y Seguridad Alimentaria para la realización del estudio.
Referencias
Referencias
Fondo de las Naciones para la Alimentación y la Agricultura, “El estado mundial de la pesca y la acuicultura 2020. La sostenibilidad en acción”, 2020. https://www.fao.org/3/ca9229es/ca9229es.pdf. [Último acceso: 07 03 2022].
P. van der Sleen et al., “Interannual temperature variability is a principal driver of low-frequency fluctuations in marine fish populations”, Commun. Biol., vol. 5, no. 1, pp. 1-8, 2022. DOI: 10.1038/s42003-021-02960-y
ComexPerú-Sociedad de Comercio Exterior del Perú, “Exportaciones acuícolas crecieron un 25,3% en el período enero-octubre de 2021”, 2022. https://www.comexperu.org.pe/articulo/exportaciones-acuicolas-crecieron-un-253-en-el-periodo-enero-octubre-de-2021. (Último acceso: 08 03 2022).
D. Schar, E. Klein, R. Laxminarayan, M. Gilbert, and T. Van Boeckel, “Global trends in antimicrobial use in aquaculture”, Sci. Rep., vol. 10, no. 1, pp. 1-9, 2020. DOI:10.1038/s41598-020-78849-3.
F. M. Treiber and H. Beranek-Knauer, “Antimicrobial residues in food from animal origin—A review of the literature focusing on products collected in stores and markets worldwide”, Antibiotics, vol. 10, no. 5, pp. 1-15, 2021. DOI: 10.3390/antibiotics10050534.
D. Schar, C. Zhao, Y. Wang, D. G. J. Larsson, M. Gilbert, and T. P. Van Boeckel, “Twenty-year trends in antimicrobial resistance from aquaculture and fisheries in Asia”, Nat. Commun., vol. 12, no. 1, pp. 1-10, 2021. DOI: 10.1038/s41467-021-25655-8. PMID: 34508079.
Administrative Committee of the Federal Register, “Electronic Code of Federal Regulations. Title 21 Food and Drugs. Part 556. Tolerances For Residues Of New Animal Drugs In Food”, 2022. https://www.ecfr.gov/cgi-bin/text-idx?SID=45a74e7727c3e71d2325986fd8064238&mc=true&tpl=/ecfrbrowse/Title21/21cfr556_main_02.tpl. [Último acceso: 15 02 2022].
H. N. Jung, D. H. Park, J. Y. Choi, S. H. Kang, H. J. Cho, J. M. Choi, J. H. Shim, A. A. Zaky, A. M. Abd El-Aty, and H. C. Shin, “Simultaneous Quantification of Chloramphenicol, Thiamphenicol, Florfenicol, and Florfenicol Amine in Animal and Aquaculture Products Using Liquid Chromatography-Tandem Mass Spectrometry”, Front. Nutr., vol. 8, no. 812803, pp. 1-10, 2022. DOI: 10.3389/fnut.2021.812803.
T. J. Centner and L. Petetin, “Divergent Approaches Regulating Beta Agonists and Cloning of Animals for Food: USA and European Union”, Society & Animals, vol. 28, no. 5-6 pp. 613-32, 2020. DOI:10.1163/15685306-12341567.
Fondo de las Naciones para la Alimentación y la Agricultura-Organización Mundial de la Salud, “Codex Alimentarius. CX/MRL 2-2018. Límites Máximos de Residuos (LMR) y recomendaciones sobre la gestión de riesgos (RGR) para residuos de medicamentos veterinarios en los alimentos”, 2018. https://www.fao.org/fao-who-codexalimentarius/sh-proxy/es/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXM%2B2%252FMRL2s.pdf. [Último acceso: 15 02 2022].
Official Journal of the European Union. “Commission Implementing Regulation (EU) 2021/808 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to be used for sampling and repealing Decisions 2002/657/EC and 98/179/EC”, 2021. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32021R0808. [Último acceso: 08 03 2022].
FDA Foods Program Regulatory Science Steering Committee, “Guidelines for the Validation of Chemical Methods in Food, Feed, Cosmetics, and Veterinary Products”, Fda.gov, 2019. https://www.fda.gov/media/81810/download. [Ultimo acceso: 08 03 2022].
Fondo de las Naciones para la Alimentación y la Agricultura-Organización Mundial de la Salud, “CAC/GL 71-2009. Adoptadas en 2009. Revisadas en 2012, 2014. Directrices para el diseño y la implementación de programas nacionales reglamentarios de aseguramiento de inocuidad alimentaria relacionados con el uso de medicamentos veterinarios en los animales destinados a la producción de alimentos”, 2014. https://www.fao.org/fileadmin/user_upload/livestockgov/documents/CXG_071s.pdf. [Último acceso: 15 02 2022].
S. Dugheri, N. Mucci, G. Cappelli, L. Trevisani, A. Bonari, E. Bucaletti, D. Squillaci, and G. Arcangeli, “Advanced Solid-Phase Microextraction Techniques and Related Automation: A Review of Commercially Available Technologies”, J. Anal. Methods Chem., vol. 2022, pp.1-15, 2022. DOI: 10.1155/2022/8690569.
B. Magnusson and U. Örnemark, “The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics”, 2014. ISBN 978-91-87461-59-0. www.eurachem.org. [Último acceso: 15 02 2022].
International Organization for Standardization / International Electrotechnical Commission, “ISO/IEC GUIDE 98-3:2008. Uncertainty of measurement — Part 3: Guide to the expression of uncertainty in measurement (GUM:1995)”, 1995. https://isotc.iso.org/livelink/livelink/fetch/2000/2122/4230450/8389141/ISO_IEC_Guide_98%2D3_2008%28E%29_%2D_Uncertainty_of_measurement_%2D%2D_Part_3%2C_Guide_to_the_expression_of_uncertainty_in_measurement_%28GUM%2C1995%29.pdf?nodeid=8389142&vernum=-2. [Último acceso: 15 02 2022].
S. L. R. Ellison and A. Williams, “EURACHEM/CITAC Guide. Quantifying Uncertainty in Analytical Measurement”, 2012. https://www.eurachem.org/images/stories/Guides/pdf/QUAM2012_P1.pdf. [Último acceso: 15 02 2022].
C. Cháfer-Pericás, Á. Maquieira, R. Puchades, B. Company, J. Miralles, and A. Moreno, “Multiresidue determination of antibiotics in feed and fish samples for food safety evaluation. Comparison of immunoassay vs LC-MS-MS”, Aquac. Res., vol. 22, no. 6, pp. 993-99, 2011. DOI: 10.1016/j.foodcont.2010.12.008.
A. Juan-García, G. Font, and Y. Picó, “Simultaneous determination of different classes of antibiotics in fish and livestock by CE-MS”, Electrophoresis, vol. 28, no. 22, pp. 4180-91, 2007. DOI: 10.1002/elps.200700383.
M. E. Dasenaki and N. S. Thomaidis, “Multi-residue determination of seventeen sulfonamides and five tetracyclines in fish tissue using a multi-stage LC-ESI-MS/MS approach based on advanced mass spectrometric techniques”, Anal. Chim. Acta, vol. 672, no. 1-2, pp. 93-102, 2010. DOI: 10.1016/j.aca.2010.04.034. Epub 2010 Apr 24. PMID: 20579496.
R. Pereira Lopes, R. Cazorla Reyes, R. Romero-González, J. L. Martínez Vidal, and A. Garrido Frenich, “Multiresidue determination of veterinary drugs in aquaculture fish samples by ultra-high performance liquid chromatography coupled to tandem mass spectrometry”, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., vol. 895-896, pp. 39-47, 2012. DOI: 10.1016/j.jchromb.2012.03.011.
B. Toussaint, M. Chedin, G. Bordin, and A. R. Rodriguez, “Determination of (fluoro)quinolone antibiotic residues in pig kidney using liquid chromatography–tandem mass spectrometry: I. Laboratory-validated method”, J. Chromatogr. A., vol. 1088, no. 1-2, pp. 32-39, 2005. DOI: 10.1016/j.chroma.2005.02.057.
M. P. Hermo, D. Barron, and J. Barbosa, “Determination of multiresidue quinolones regulated by the European Union in pig liver samples. High-resolution time-of-flight mass spectrometry versus tandem mass spectrometry detection”, J. Chromatogr. A., vol. 1201, no. 1, pp. 1-14, 2008. DOI: 10.1016/j.chroma.2008.05.090.
G. van Vyncht, A. Janosi, G. Bordin, B. Toussaint, G. Maghuin-Rogister, E. De Pauw, and A. R. Rodriguez, “Multiresidue determination of (fluoro)quinolone antibiotics in swine kidney using liquid chromatography– tandem mass spectrometry”, J. Chromatogr. A., vol. 952, no. 1-2, pp.121-29, 2002. DOI: 10.1016/s0021-9673(02)00092-4.
P. Mottier, Y. A. Hammel, E. Gremaud, and P. A. Guy, “Quantitative high-throughput analysis of 16 (fluoro)quinolones in honey using automated extraction by turbulent flow chromatography coupled to liquid chromatograph- tandem mass spectrometry”, J. Agric. Food Chem., vol. 56, no. 1, pp. 35-43, 2008. DOI: 10.1021/jf072934d.
S. Bogialli, G. D’Ascenzo, A. Di Corcia, A. Lagana, and G. Tramontana, “Simple assay for monitoring seven quinolone antibacterials in eggs: Extraction with hot water and liquid chromatography coupled to tandem mass spectrometry Laboratory validation in line with the European Union Commission Decision 657/2002/EC”, J. Chromatogr. A., vol. 1216, no. 5, pp. 794-780, 2009. DOI: 10.1016/j.chroma.2008.11.070.
Cómo citar
IEEE
ACM
ACS
APA
ABNT
Chicago
Harvard
MLA
Turabian
Vancouver
Descargar cita
Licencia
Derechos de autor 2023 Diego Chirinos Pajuelo, Orlando Lucas Aguirre, Wilfredo León Gonzales, Nathaly Elizabeth Hurtado Galindo, Estefania Morales Ochante, Luis Huicho, Maria Concepcion Rivera Chira

Esta obra está bajo una licencia internacional Creative Commons Atribución 4.0.
Los autores/as conservarán sus derechos de autor y garantizarán a la revista el derecho de primera publicación de su obra, el cuál estará simultáneamente sujeto a la Licencia de reconocimiento de Creative Commons (CC. Atribución 4.0) que permite a terceros compartir la obra siempre que se indique su autor y su primera publicación en esta revista.
Los autores/as podrán adoptar otros acuerdos de licencia no exclusiva de distribución de la versión de la obra publicada (p. ej.: depositarla en un archivo telemático institucional o publicarla en un volumen monográfico) siempre que se indique la publicación inicial en esta revista.
Se permite y recomienda a los autores/as difundir su obra a través de Internet (p. ej.: en archivos telemáticos institucionales o en su página web) antes y durante el proceso de envío, lo cual puede producir intercambios interesantes y aumentar las citas de la obra publicada. (Véase El efecto del acceso abierto).










